Download to read offline



![Control Structures
if condition:
statements
[elif condition:
statements] ...
else:
statements
while condition:
statements
for var in sequence:
statements
break
continue](https://image.slidesharecdn.com/p72018biopython3-181203231826/85/P7-2018-biopython3-5-320.jpg)
![Lists
• Flexible arrays, not Lisp-like linked
lists
• a = [99, "bottles of beer", ["on", "the",
"wall"]]
• Same operators as for strings
• a+b, a*3, a[0], a[-1], a[1:], len(a)
• Item and slice assignment
• a[0] = 98
• a[1:2] = ["bottles", "of", "beer"]
-> [98, "bottles", "of", "beer", ["on", "the", "wall"]]
• del a[-1] # -> [98, "bottles", "of", "beer"]](https://image.slidesharecdn.com/p72018biopython3-181203231826/85/P7-2018-biopython3-6-320.jpg)
![Dictionaries
• Hash tables, "associative arrays"
• d = {"duck": "eend", "water": "water"}
• Lookup:
• d["duck"] -> "eend"
• d["back"] # raises KeyError exception
• Delete, insert, overwrite:
• del d["water"] # {"duck": "eend", "back": "rug"}
• d["back"] = "rug" # {"duck": "eend", "back":
"rug"}
• d["duck"] = "duik" # {"duck": "duik", "back":
"rug"}](https://image.slidesharecdn.com/p72018biopython3-181203231826/85/P7-2018-biopython3-7-320.jpg)



























![Numpy – SciPy – Matplib -- > PANDAS ?
• Python Data Analysis Library, similar to:
– R
– MATLAB
– SAS
• Combined with the IPython toolkit
• Built on top of NumPy, SciPy, to some extent
matplotlib
• Panel Data System
– Open source, BSD-licensed
• Key Components
– Series is a named Python list (dict with list as value).
{ ‘grades’ : [50,90,100,45] }
– DataFrame is a dictionary of Series (dict of series):
{ { ‘names’ : [‘bob’,’ken’,’art’,’joe’]}
{ ‘grades’ : [50,90,100,45] }
}](https://image.slidesharecdn.com/p72018biopython3-181203231826/85/P7-2018-biopython3-38-320.jpg)





![Calculation of Propensities
Pr[i|b-sheet]/Pr[i], Pr[i|-helix]/Pr[i], Pr[i|other]/Pr[i]
determine the probability that amino acid i is in
each structure, normalized by the background
probability that i occurs at all.
Example.
let's say that there are 20,000 amino acids in the database, of
which 2000 are serine, and there are 5000 amino acids in
helical conformation, of which 500 are serine. Then the
helical propensity for serine is: (500/5000) / (2000/20000) =
1.0](https://image.slidesharecdn.com/p72018biopython3-181203231826/85/P7-2018-biopython3-44-320.jpg)

![Calculation of Propensities
Pr[i|b-sheet]/Pr[i], Pr[i|-helix]/Pr[i], Pr[i|other]/Pr[i]
determine the probability that amino acid i is in
each structure, normalized by the background
probability that i occurs at all.
Example.
let's say that there are 20,000 amino acids in the database, of
which 2000 are serine, and there are 5000 amino acids in
helical conformation, of which 500 are serine. Then the
helical propensity for serine is: (500/5000) / (2000/20000) =
1.0](https://image.slidesharecdn.com/p72018biopython3-181203231826/85/P7-2018-biopython3-46-320.jpg)

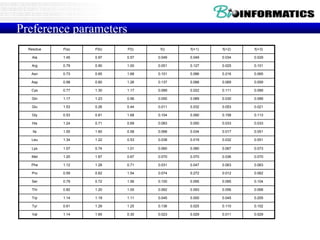




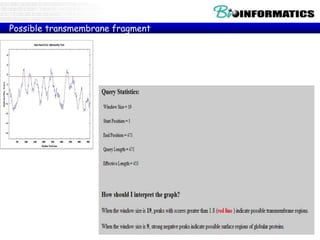
The document discusses various bioinformatics tools and algorithms for analyzing protein sequences, including Biopython for working with biological sequence data, the Kyte-Doolittle algorithm for predicting transmembrane regions, and the Chou-Fasman algorithm for predicting secondary structure from amino acid preferences for alpha helices, beta sheets, and random coils. It also provides examples of analyzing Swiss-Prot data to find properties of human proteins and applying these tools and libraries to extract insights from protein sequences.







![Bio ontologies and semantic technologies[2]](https://cdn.slidesharecdn.com/ss_thumbnails/bioontologiesandsemantictechnologies2-180509123734-thumbnail.jpg?width=640&height=640&fit=bounds)


































































