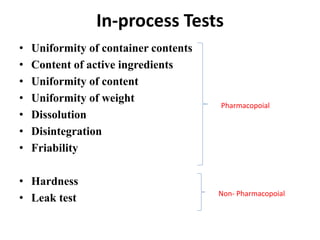
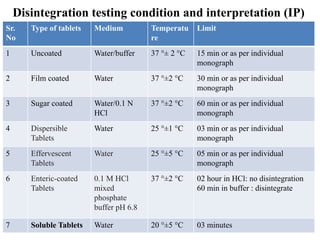
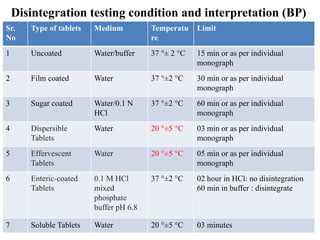
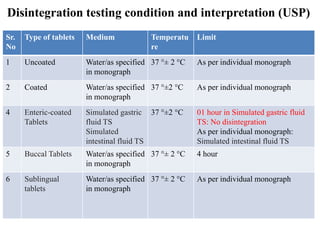
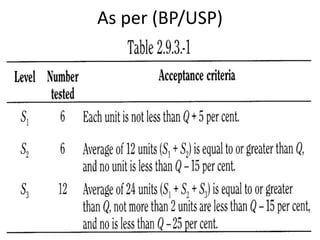
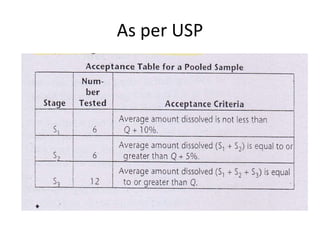
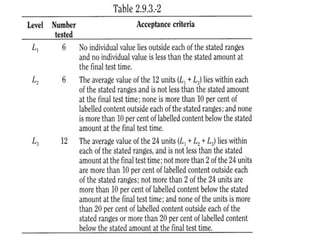
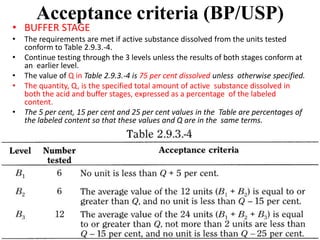
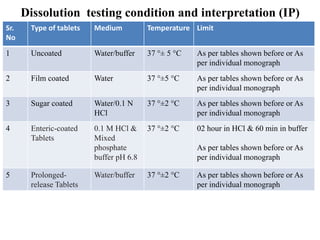
This document provides information about tablets, including their definition, categories, in-process tests, and testing methods. Tablets are solid oral dosage forms containing medicaments. There are several categories including uncoated, film coated, sugar coated, and modified release tablets. In-process tests include uniformity of contents, weight, dissolution, and disintegration. Dissolution and disintegration tests are described for different tablet types using specified apparatus, media, and time/acceptance criteria. Modified and prolonged release tablets have additional dissolution testing methods and criteria for acid and buffer stages.







































































































![CASE_PRESENTATION_ON_subdural_hematoma(SDH)[1 FINAL PPT]-1.pptx](https://cdn.slidesharecdn.com/ss_thumbnails/casepresentationonsubduralhematomasdh1finalppt-1-260129172522-d405d375-thumbnail.jpg?width=640&height=640&fit=bounds)






