Downloaded 341 times





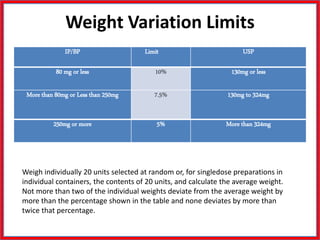



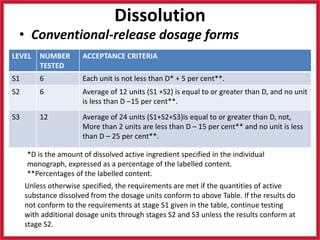





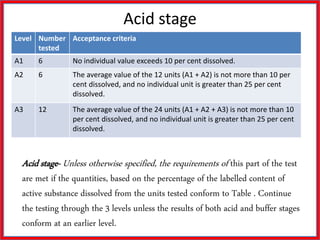


This document summarizes the standards and testing methods for different types of tablets according to the Indian Pharmacopoeia. It describes 10 types of tablets and the standards that apply to all tablets, including content uniformity, weight variation, disintegration, friability, and dissolution testing. The document provides details on the acceptance criteria and testing procedures for each of these standards.


































































































