In process quality control and finished products quality control for tablets in the Pharma industry according to Indian,
US and British pharmacopoeias.
INRODUCTION
● Quality controlis vital in ensuring the safety, efficacy, and consistency of
pharmaceutical tablets.
● IPQC means controlling the procedures involved during manufacturing of the dosage
forms starting from raw material purchase to dispatch of the quality product in ideal
packaging.
● In process material should be tested for identity, strength, quality and purity as
appropriate and approved or rejected by the quality control unit during the production
process.
3.
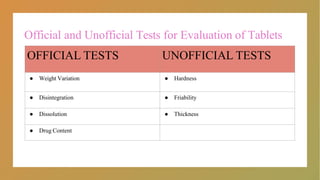
Official and UnofficialTests for Evaluation of Tablets
OFFICIAL TESTS UNOFFICIAL TESTS
● Weight Variation ● Hardness
● Disintegration ● Friability
● Dissolution ● Thickness
● Drug Content
4.
1. Weight Variation
□The weight of the tablet is the quantity of the granulation that contains the labeled
amount of the therapeutic ingredients.
□After the tablet machine in the operation the weight of tablet is routinely checked to
ensure that proper tablet weights are made.
PROCEDURE:
□Weight 20 tablet selected at random , each one individually.X1,X2,X3…..Xz
□To determine average weight. X –(X1+X2+X3…+Xz)/20
5.
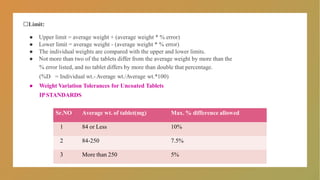
□Limit:
● Upper limit= average weight + (average weight * % error)
● Lower limit = average weight - (average weight * % error)
● The individual weights are compared with the upper and lower limits.
● Not more than two of the tablets differ from the average weight by more than the
% error listed, and no tablet differs by more than double that percentage.
(%D = Individual wt.-Average wt./Average wt.*100)
● Weight Variation Tolerances for Uncoated Tablets
IPSTANDARDS
Sr.NO Average wt. of tablet(mg) Max. % difference allowed
1 84 or Less 10%
2 84-250 7.5%
3 More than 250 5%
6.
ACCEPTANCE CRITERIA
● Productcomplies the test if not more than two of the individual
weights deviate from the average weight by more than the percentage
shown in the above table and none deviates by more than twice that
percentage.
7.
2. Drug content
➢This test is done to ensure that every tablet contain the amount of drug
substance intended with little variation within a batch.
PROCEDURE
➢ Randomly select 30 tablets. 10 of these assayed individually.
➢ The Tablet pass the test if 9 of the 10 tablets must contain not less than 85
% and not more than 115 % of the labeled.
➢ Drug content and the 10th tablet may not contain less than 75 % and more
than 125 % of the labeled content.
8.
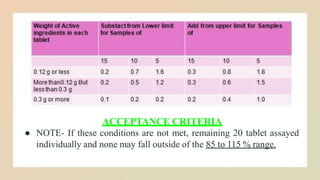
ACCEPTANCE CRITERIA
● NOTE-If these conditions are not met, remaining 20 tablet assayed
individually and none may fall outside of the 85 to 115 % range.
9.
3. Disintegration Test
□It is the time required for tablet to break into particles,
□ The disintegration test is a measure only of time required under a given set
of conditions for a group of tablets to disintegrate into particles.
□ It is performed to identify the disintegration of tablet in particular time
period.
□ Disintegration test is not performed for controlled & sustained release tablets.
□ According to test tablet must disintegrate and all particles must pass through
the 10 mesh screen in the time specified.
□ If any residue remains, it must have a soft mass.
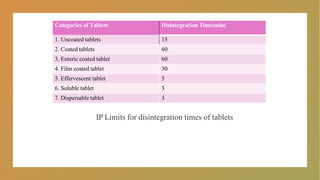
Categories of TabletsDisintegration Time(min)
1. Uncoated tablets 15
2. Coated tablets 60
3. Enteric coated tablet 60
4. Film coated tablet 30
5. Effervescent tablet 5
6. Soluble tablet 3
7. Dispersable tablet 3
IP Limits for disintegration times of tablets
12.
ACCEPTANCE CRITERIA
● Tabletspass the test if all 6 of them have disintegrated .
● If 1 or 2 fail to disintegrate,repeat the test on additional 12 tablets;
and now 16 of the total 18 tablets must disintegrate for the product to
comply the test.
● If the tablet adhere to the disc and the preparation fails to comply
,repeat the test omitting the disc. The preparation complies the test if
all the tablets in the repeat test disintegrate.
13.
4.Dissolution Test
□Dissolution isperformed to check the percentage release from the dosage forms.
i.e. Tablet.
□ Tablet breaks down into small particles which offers a greater surface area to the
dissolving media.
□ Disintegration test does not give assurance that particles will release drug in solution at
an appropriate rate, that’s why dissolution tests & its specifications developed for all
tablet products.
14.
□
Dissolution test apparatusIP apparatus
1 First is Paddle apparatus (IP)
2 Second is Basket apparatus (IP)
1.First is Paddle apparatus (IP): Immediate release tablet (conventional
tablet)
1.Dissolution apparatus – Type 1 and Type 2
2.Temperature - 37±0.5˚C
3.Time – 30 min
4. Time of interval – 5, 10, 15, 20, 25, 30.
5. Media – PH 1.2Acidic Buffer, PH 4.5Acetate buffer, PH 5.8 Phosphate buffer.
6. Rpm – 75 -100 rpm
7.Volume – 900 ml
Procedure: The tablet was added into cylindrical vessel containing 1000 ml dissolution
media having rpm 75 and tem.37±0.5˚C. Dissolution of tablet was conducted 30 min, in
5 min. of interval, after 5 min 5 mL sample was removed and appropriate quantity of
sample take absorbance by using U.V. spectroscopy technique and determine rate of
dissolution of tablet.
15.
□Second is Basketapparatus (IP)
Sustained release tablet
1.Dissolution apparatus – Type 2
2.Temperature - 37±0.5˚C
3.Time – 7 hrs
4.Media – PH 1.2Acidic Buffer, PH 6.8 Phosphate buffer.
5.Rpm – 75 – 100.
6.Volume – 900 ml.
Procedure - The tablet was added into cylindrical vessel containing 1000 ml
PH 1.2 Acidic media having rpm 75 for next two hours and tem. 37±0.5˚C.
Dissolution media was changes tablet was added in to PH 6.8 Phosphate buffer
for next five hour for 1 hr. of interval. After 1 hr. 5 mL sample was removed
and appropriate quantity of sample take absorbance by using U.V.
spectroscopy technique and determine rate of dissolution of tablet.
16.
Sr.no. Quantity
Stage/level
Number of
tabletstested
Acceptance criteria
1 S1 6 Each unit is < D* + 5 percent**
2 S2 6 Average of 12 units (S1 +S2) is equal to or greater
than (> )D, and no unit is less than D - 15 percent**
3 S3 12 Average of 24 units (S1+S2+S3) is equal to or
greater than (> )D, not more than 2 units are less
than d-15 percent** and no unit is less than d-25 percent**
*D is the amount of dissolved active ingredient specified in the individual monograph, expressed as a
percentage of the labelled content.
** Percentages of the labelled content.
ACCEPTANCE CRITERIA
17.
1.Hardness
● Tablet requiresa certain amount of strength or hardness and resistance to friability to withstand
mechanical shocks of handling in manufacture, packaging and shipping. Hardness generally
measures the tablet crushing strength.
FactorsAffecting the Hardness:
than direct
➢ Compression of the tablet and compressive force.
➢ Amount of binder. (More binder à more hardness)
➢ Method of granulation in preparing the tablet (wet method gives more hardness
method, Slugging method gives the best hardness).
18.
20
● Some ofdevices used to test tablet
hardness are:
1. Monsanto
2. Pfizer
3. Schleuniger
Oral tablets 4-10 kg
Chewable tablets 3 kg
Sustained release tablets 10-20 kg
● LIMITS: (Take 5 tablets and avg. out)
19.
2.Friability
● Friability ofa tablet can determine in laboratory by Roche friabilator. This consist of a plastic
chamber that revolves at 25 rpm, dropping the tablets through a Distance of six inches in the
friabilator, which is then operate for 100 revolutions.
PROCEDURE:
➢ Weight 20 tablets altogether -w1.
➢ Put these tablet in the friabilator and adjust instrument at
100 rpm.
➢ Weigh the 20 tablets -w2.
➢ Friability(%loss) –It must or less than or equal to 1%.
20.
ACCEPTANCE CRITERIA
● Amaximum loss of weight not greater than 1.0% is acceptable for
most tablets.
● If cracked, chipped or broken tablets are present in the sample after
tumbling,it’s obvious that the sample fails the test.
21.
3.Thickness
➢ The thicknessof a tablet is the only dimensional variable related
to the process.
➢ Thickness of individual tablets may be measured by a Vernier
calliper.
➢ Other techniques involve placing 5 or 10 tablets in a holding tray,
where their total thickness may be measured by a sliding caliper
scale.
➢ Tablet thickness should be controlled within a ± 5 % variation of
a standard.
➢ Thickness must be controlled to facilitate packaging. It is
expressed in mm.
22.
REFRENCES
1.Comparitive study ofIn-process and Finished Products Quality control Tests of IP,BP, & USP for
tablets International journal of Pharmacy Teeaching & Practices 2011,vol.2,issue 4,176-183.
2. Indian Pharmacopoeia-2010.
3. Leon Lachman, The theory and practice of Industrial pharmacy,3rd edition ,page no
-67-68,315-317,296-303.