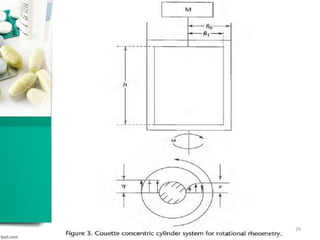
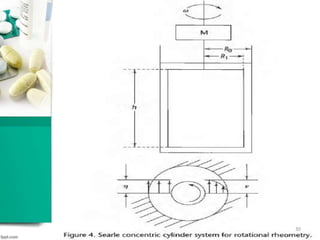
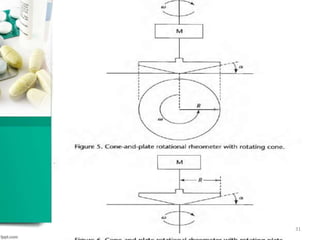
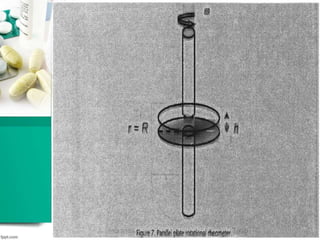
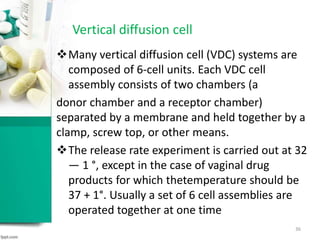
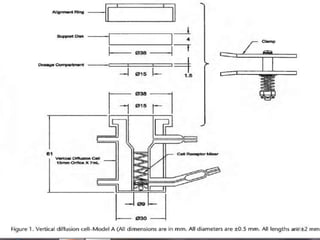
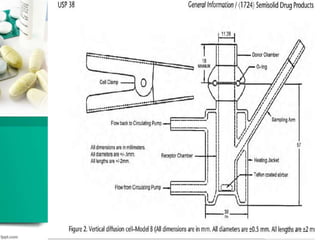
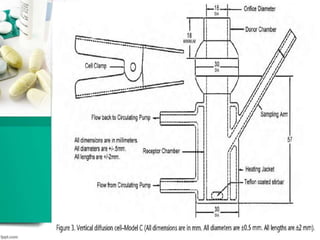
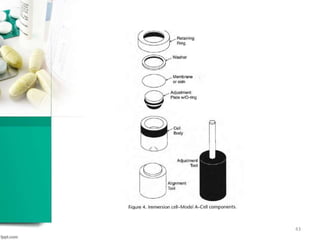
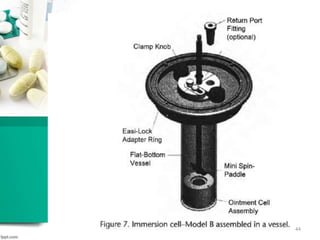
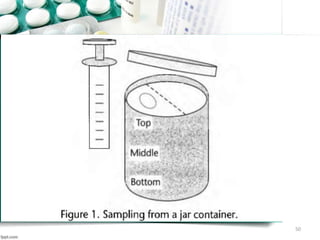
This document discusses various quality control tests for semisolid products like lidocaine ointment. It describes tests such as infrared absorption, assay testing using HPLC, uniformity of dosage, preservative effectiveness testing, pH testing, and viscosity testing. The infrared absorption test verifies the identity of lidocaine in the ointment. The assay test quantifies the amount of lidocaine using HPLC. Tests like uniformity of dosage and preservative effectiveness ensure consistency and prevent microbial growth.





![Lidocaine OintmentASSAY
• Procedure
Solution A: 0.1% phosphoric acid, prepared by adding
1.0 mL of 85% phosphoric acid to 1 L of water.
Solution B: Acetonitrile
Diluent: Acetonitrile and Solution A (1:1)
System suitability solution: 0.1 mg/mL of USP Lidocaine
RS and 0.04 mg/mL of USP Ropivacaine Related
Compound A RS in Diluent
[Note—USP Ropivacaine Related Compound A RS is
2 , 6 -dimethylaniline hydrochloride.]
Standard solution: 0.1 mg/mL of USP Lidocaine RS in
Diluent
Sample solution: Nominally 0.1 mg/mL of lidocaine in
Diluent from a portion of Ointment. Sonicate the solution
for about 5 min.
Chromatographic system
(See Chromatography (621), System Suitability.)
Mode: LC
Detector: UV 210 nm
Column: 4.6-mm x 15-cm; 5-jjjti packing L1
Flow rate: 0.8 mL/min
Injection volume: 5 jiL
System suitability
Samples: System suitability solution and Standard
solution
[Note—The relative retention times for 2,6-dimethylaniline 5](https://image.slidesharecdn.com/qcsemisolidmaryamkazemi-160827144143/85/Quality-control-of-semisolids-5-320.jpg)
![.
and lidocaine are about 0.93 and 1.0,
respectively.]
Suitability requirements
Tailing factor: NMT 1.5 for the lidocaine peak, System
suitability solution
Relative standard deviation: NMT 2.0%, Standard
solution
Resolution: NLT 1.8 between lidocaine and 2,6-
dimethylaniline, System suitability solution
Analysis
Samples: Standard solution and Sample solution
Calculate the percentage of the labeled amount of lidocaine
(C14H22N2O) in the portion of Ointment taken:
Result = (ru//s) x (Cs/Cu) x 100
ru = peak response from the Sample solution
rs = peak response from the Standard solution
Q = concentration of USP Lidocaine RS in the
Standard solution (mg/mL)
Cu = nominal concentration of lidocaine in the
Sample solution (mg/mL)
Acceptance criteria: 95.0%-105.0%
6](https://image.slidesharecdn.com/qcsemisolidmaryamkazemi-160827144143/85/Quality-control-of-semisolids-6-320.jpg)


![Solid, Semi-Solid, and Liquid Dosage Forms
The requirements for dosage uniformity are met if the
acceptance value of the first 10 dosage units is less
than or equal to
L1%. If the acceptance value is > L1%, test the next 20
units, and calculate the acceptance value. The
requirements are met if
the final acceptance value of the 30 dosage units is <
LI %, and no individual content of ‘any, dosage unit is
less than [1 -
(0.01)(L2)]M nor more than [1 + (0.01)(L2)]M ‘ as
specified, in the Calculation of Acceptance Value
under Content Uniformity or
under *Weight4 Variation. Unless otherwise specified,
LI is 15.0 and L2 is 25.
9](https://image.slidesharecdn.com/qcsemisolidmaryamkazemi-160827144143/85/Quality-control-of-semisolids-9-320.jpg)