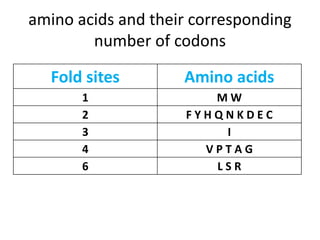
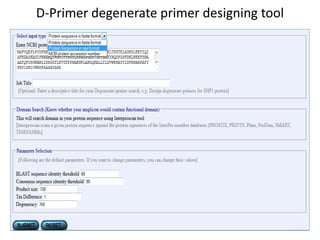
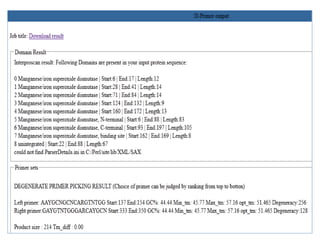
Degenerate primers are designed from conserved amino acid sequences aligned from multiple species. They contain nucleotide degeneracies that allow binding to related gene sequences. Key steps are:
1) Identifying conserved regions over 5+ amino acids long and within 200-600 bp for primers.
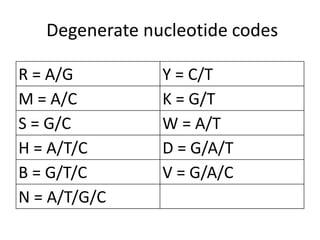
2) Calculating primer degeneracy based on contributing amino acids to minimize values over 64.
3) Optimizing primers by avoiding amino acids with 6-fold degeneracy and adding 5' tails.




















































![PCR and Primer design[604].pdf](https://cdn.slidesharecdn.com/ss_thumbnails/pcrandprimerdesign604-230611034504-10d9745b-thumbnail.jpg?width=640&height=640&fit=bounds)

























![谷歌留痕技术 [ 𝙩𝙤𝙥 𝟮𝟯𝟯. 𝙘 𝙤𝙢 ]](https://cdn.slidesharecdn.com/ss_thumbnails/top233-260130174328-3833018c-thumbnail.jpg?width=640&height=640&fit=bounds)


















