Downloaded 131 times







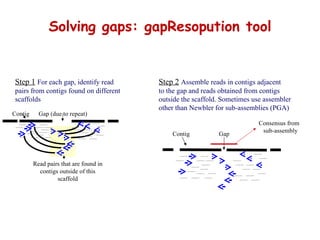
![Solving gaps: gapResopution tool (II) Step 3 If gap is not closed, tool designs designs primers for sequencing reactions Step 4 Iterate as necessary (in sub-assemblies) http://www.jgi.doe.gov/ [email_address] Contig Gap Design sequencing reactions to close gap](https://image.slidesharecdn.com/assemblyandfinishing-120101035409-phpapp01/85/Assembly-and-finishing-19-320.jpg)

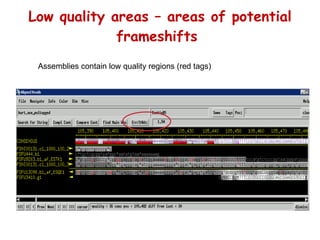
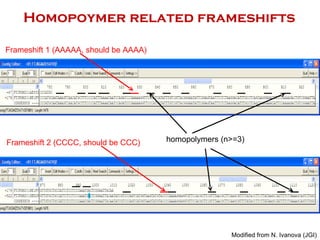
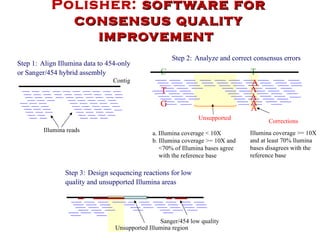


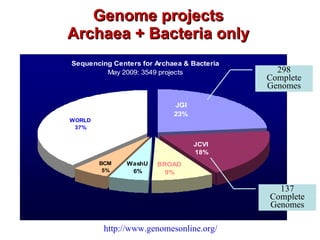

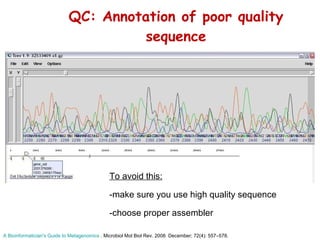
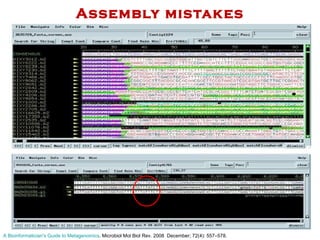

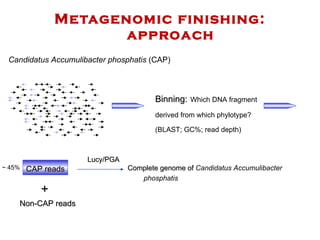
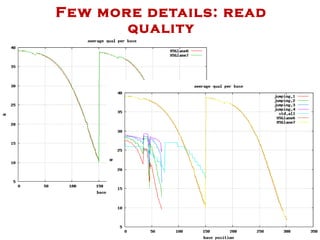


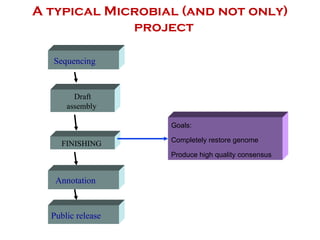
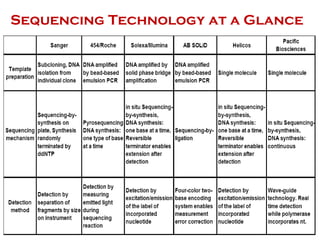
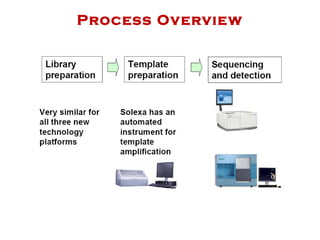
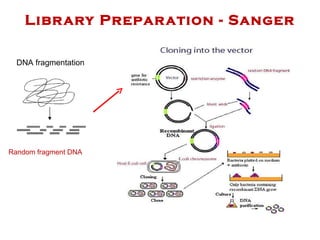
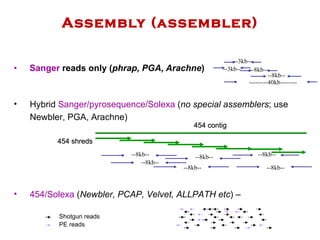
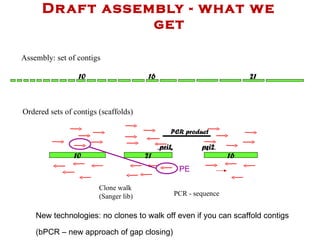
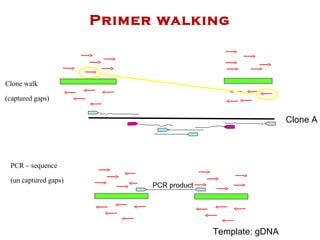
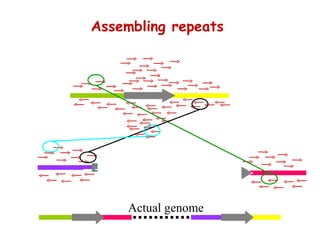
The document discusses genome assembly and finishing processes. It begins by outlining typical project goals of completely restoring the genome and producing a high-quality consensus sequence. It then describes the evolution of sequencing technologies from Sanger to newer platforms and their impact on draft assemblies. Key steps in the assembly and finishing process include library preparation, assembly, identifying gaps, and improving consensus quality.















































































![Getting Started with Apache Spark: Big Data Made Simple [Free Meetup]](https://cdn.slidesharecdn.com/ss_thumbnails/apachesparkgettingstarted-260203175547-8361bcc3-thumbnail.jpg?width=640&height=640&fit=bounds)







