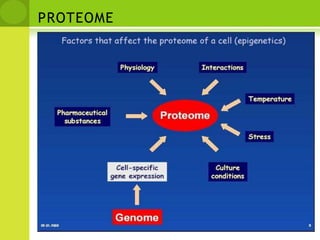
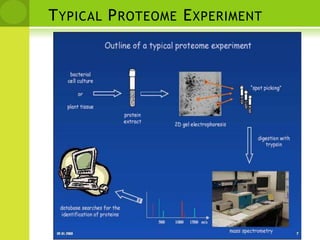
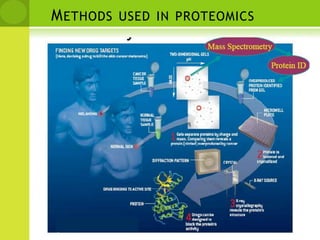
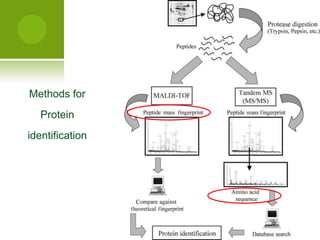
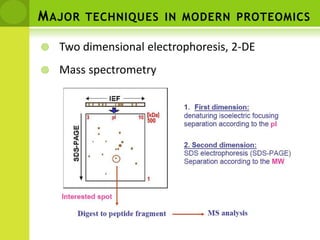
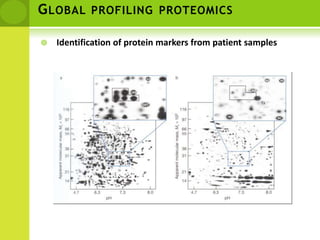
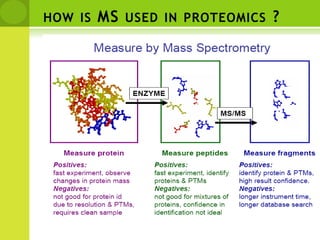
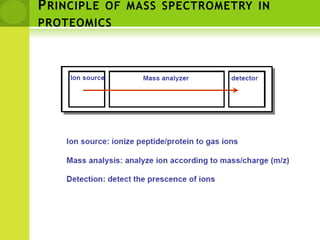
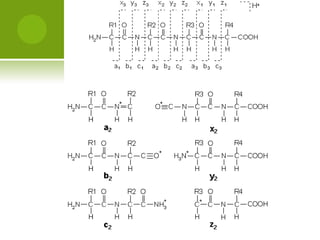
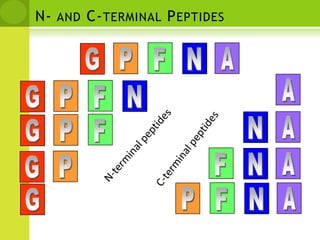
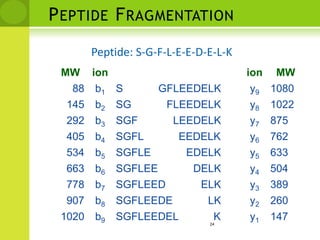
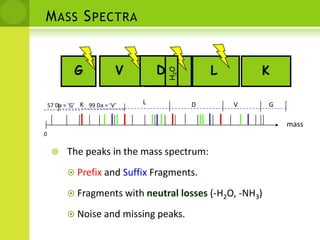
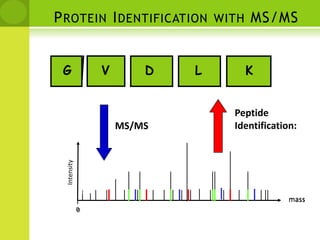
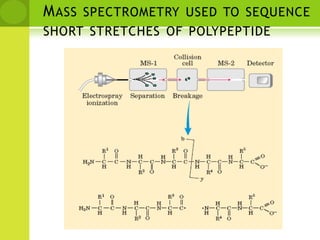
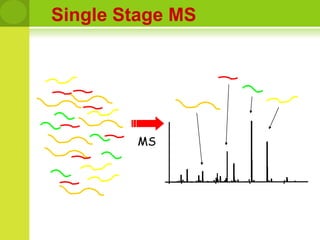
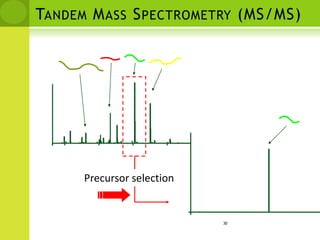
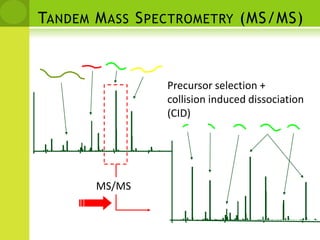
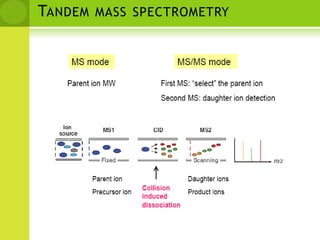
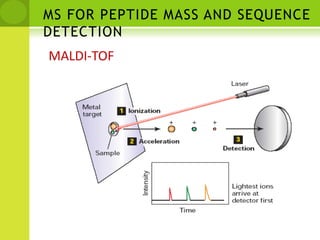
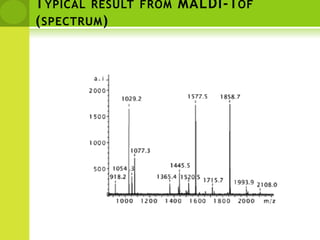
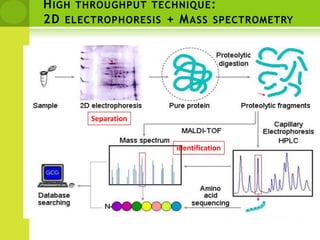
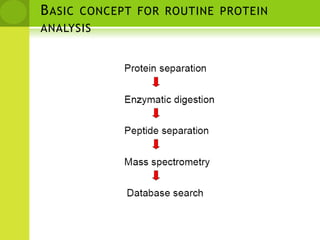
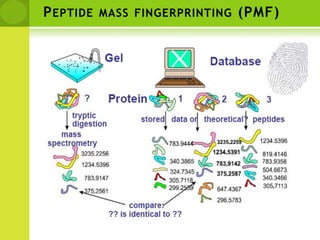
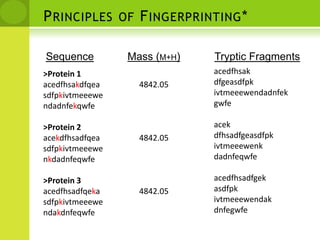
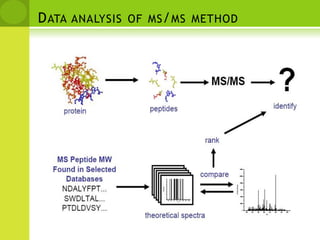
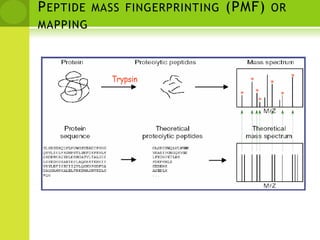
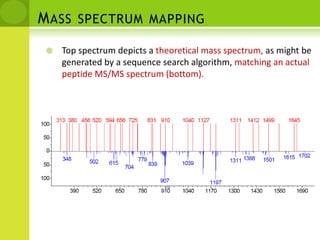
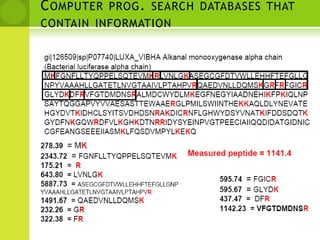
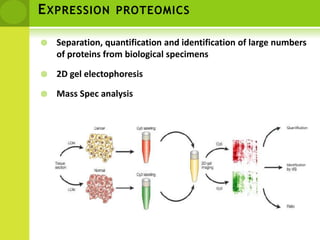
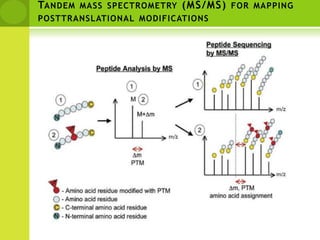
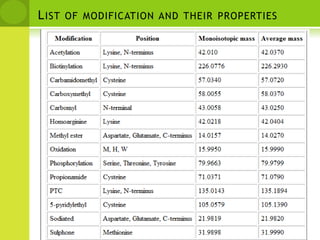
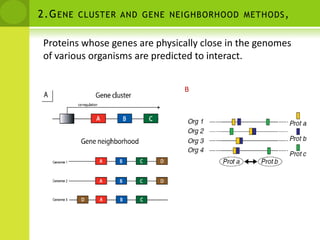
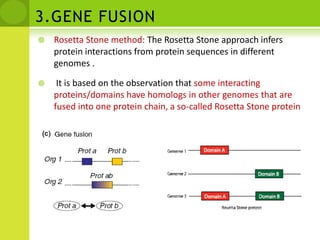
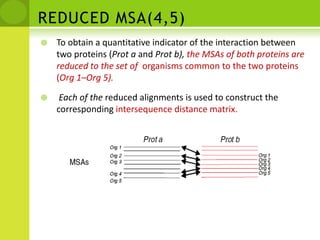
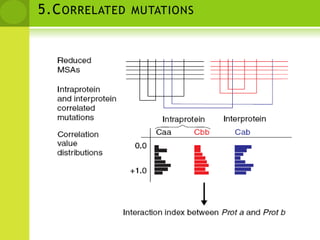
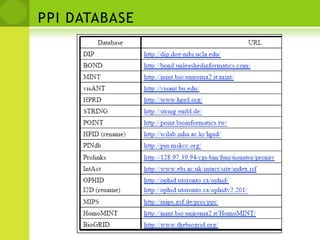
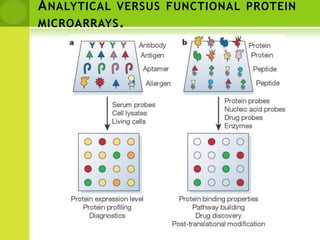
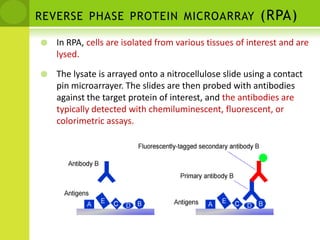
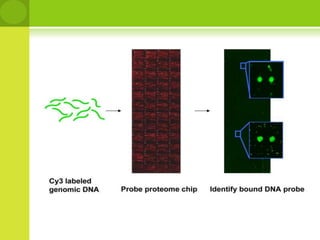
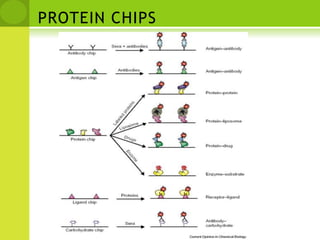
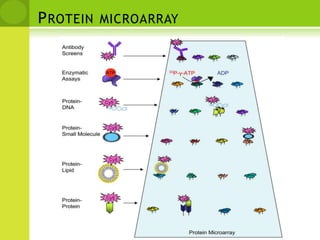
The document discusses the field of proteomics, which is the large-scale study of proteins, including their functions and structures. It defines proteomics and describes several areas within it, such as functional proteomics, expressional proteomics, and structural proteomics. It outlines typical proteomics experiments and some key methods used, including two-dimensional electrophoresis, mass spectrometry, and protein-protein interaction prediction methods like phylogenetic profiling.