Downloaded 85 times






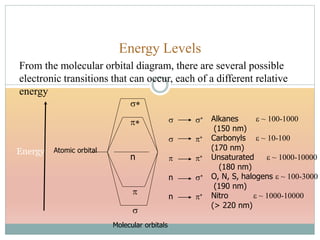
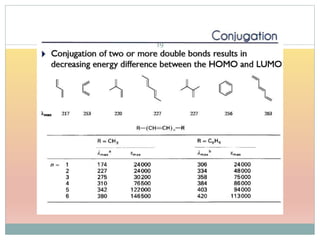
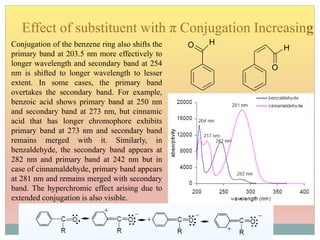
![Effect of Conjugation
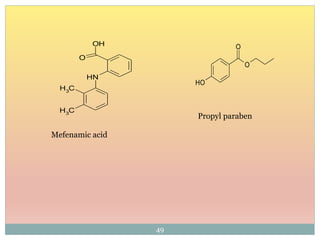
15Molecular structure or environment [Conjugation (substitution) / Auxochrome or change of
solvent]can influence λmax and ε.
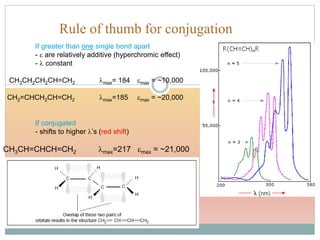
Shift to longer λ bathochromic/red e.g. Ethylene (170 nm) 1,3 –butadiene (217 nm)
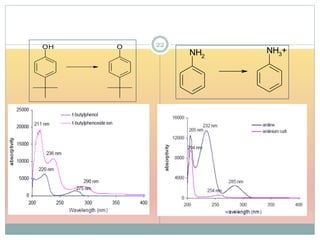
Shift to shorter λ hypsochromic/ blue e.g. aniline (285 nm) and anilinium ion (254 nm)
Increase in ε hyperchromic effect e.g. pyridine (2750) 2-methyl pyridine (3560)
Decrease in ε hypochromic effect e.g. biphenyl (6540) 3- methyl biphenyl (5970)
Due to solvent change, UV-Visible spectral changes in next slides, Solvent Effect.](https://image.slidesharecdn.com/40-191218142647/85/UV-SPECTROSCOPY-ULTRA-VIOLET-SPECTROSCOPY-15-320.jpg)
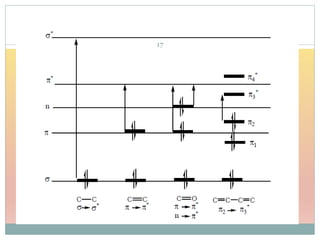




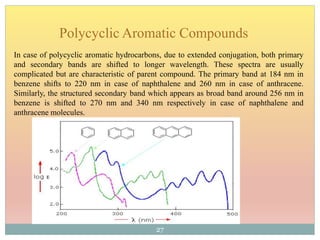

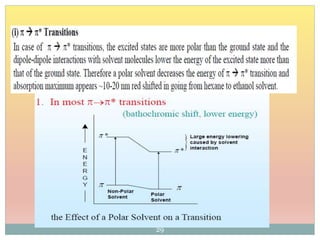
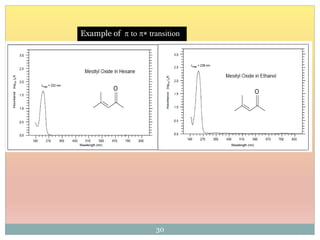

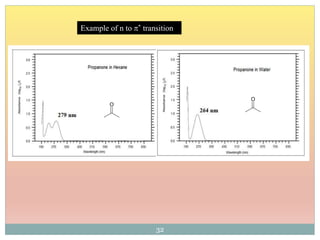
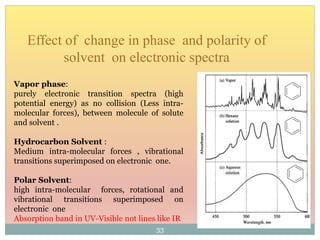
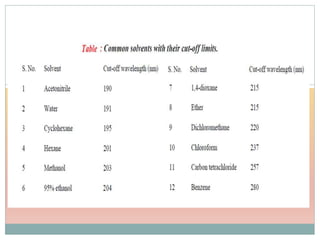

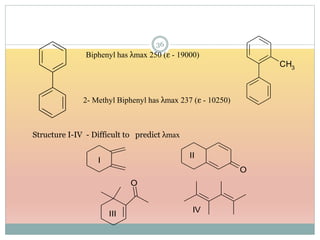
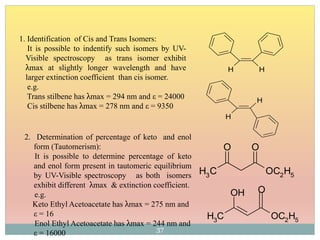



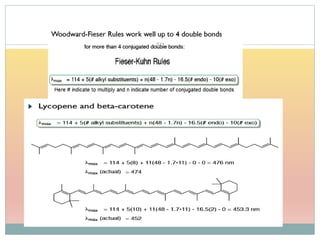

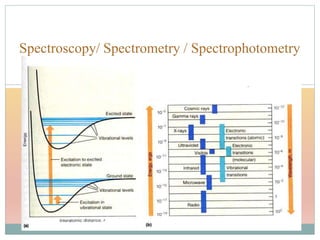
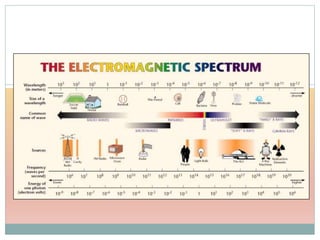
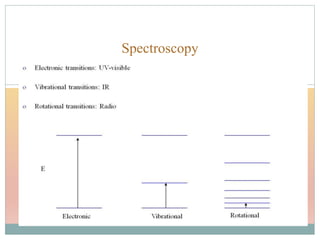
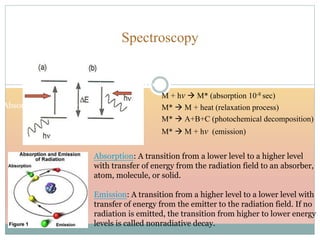
The document provides an in-depth overview of UV-Visible spectroscopy, detailing the concepts of absorption, emission, and the transitions of electrons between molecular orbitals. It discusses the roles of chromophores and auxochromes, the effects of substituents on the spectral properties of organic compounds, and selection rules governing electronic transitions. Additionally, it covers the influence of molecular structure, solvent effects, and empirical rules for predicting spectral behavior in various organic compounds.























![Noesy [autosaved]](https://cdn.slidesharecdn.com/ss_thumbnails/noesyautosaved-200728183752-thumbnail.jpg?width=640&height=640&fit=bounds)






























![problems associated with skin[ as per Pharmaceutics]](https://cdn.slidesharecdn.com/ss_thumbnails/2-200204102918-thumbnail.jpg?width=640&height=640&fit=bounds)
![cosmetics and cosmeticals [department of pharmaceutics]](https://cdn.slidesharecdn.com/ss_thumbnails/1-200204101110-thumbnail.jpg?width=640&height=640&fit=bounds)
![MUTUAL PRODRUG [PHARMACEUTICALS]](https://cdn.slidesharecdn.com/ss_thumbnails/67-191220064837-thumbnail.jpg?width=640&height=640&fit=bounds)
![TITANIUM CHLORIDE [PHARMACEUTICAL REAGENT]](https://cdn.slidesharecdn.com/ss_thumbnails/66-191220064235-thumbnail.jpg?width=640&height=640&fit=bounds)
![OLIGONUCLEOTIDE THERAPY [ TECHNIQUES, APPLICATIONS]](https://cdn.slidesharecdn.com/ss_thumbnails/66-191220063512-thumbnail.jpg?width=640&height=640&fit=bounds)
![OXIDATION [PHARMACEUTICAL PROCESS CHEMISTRY]](https://cdn.slidesharecdn.com/ss_thumbnails/65-191220062805-thumbnail.jpg?width=640&height=640&fit=bounds)

![PHASE TRANSFER CATALYSIS [PTC]](https://cdn.slidesharecdn.com/ss_thumbnails/64-191219161253-thumbnail.jpg?width=640&height=640&fit=bounds)

![LIQUID CHROMATOGRAPHY- MASS SPECTROSCOPY[LC-MS]](https://cdn.slidesharecdn.com/ss_thumbnails/63-191219160156-thumbnail.jpg?width=640&height=640&fit=bounds)

![Homogeneous catalysis [ MPHARM, MSC, BPHARM, BSC]](https://cdn.slidesharecdn.com/ss_thumbnails/62-191219155346-thumbnail.jpg?width=640&height=640&fit=bounds)
![Dicyclohexylcarbodiimide [DCC]](https://cdn.slidesharecdn.com/ss_thumbnails/61-191219150428-thumbnail.jpg?width=640&height=640&fit=bounds)




















