Downloaded 115 times










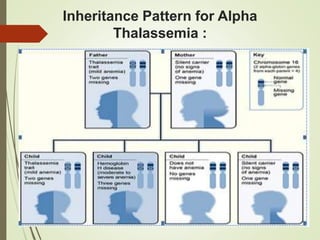


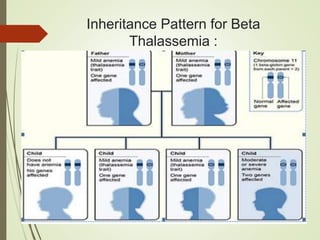

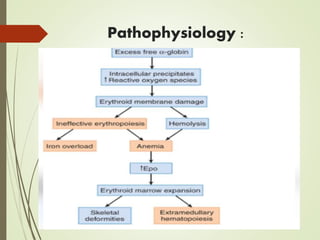
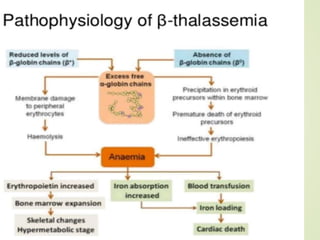

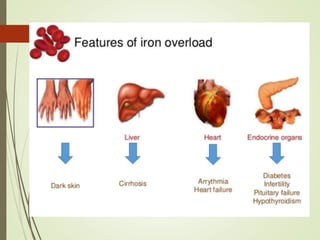


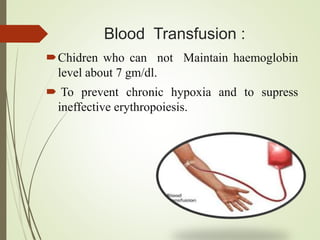








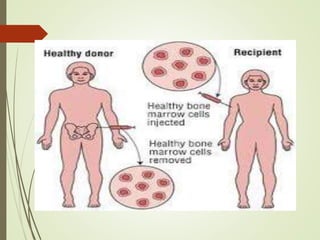
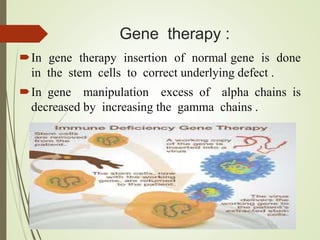









Thalassemia is an inherited blood disorder characterized by inadequate hemoglobin production due to genetic factors, leading to types such as alpha and beta thalassemia. Diagnosis includes patient history, physical examination, and various lab tests, while management may involve blood transfusions, iron chelation therapy, and potential bone marrow transplantation. Prevention strategies focus on genetic counseling and screening for carrier status in at-risk individuals planning to have children.























































































