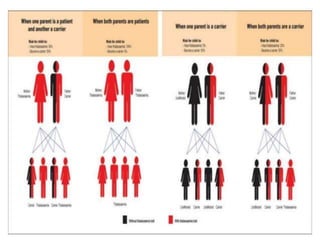
This document discusses thalassemia, a hereditary blood disorder caused by an imbalance in hemoglobin production. It describes the types of thalassemia, including alpha and beta thalassemia minor, intermedia, and major. For beta thalassemia major, regular blood transfusions are necessary to survive. Clinical manifestations include severe anemia, bone deformities, and organomegaly. Diagnosis involves blood tests and molecular analysis. Treatment depends on severity but may include frequent blood transfusions, iron chelation therapy, and bone marrow transplantation. Nursing care focuses on monitoring vital signs, managing transfusions and chelation therapy, and providing education to patients and families.