


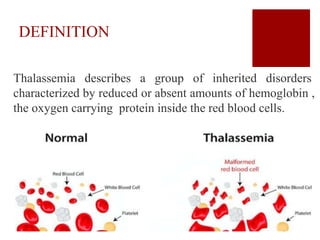

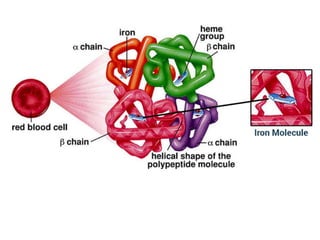





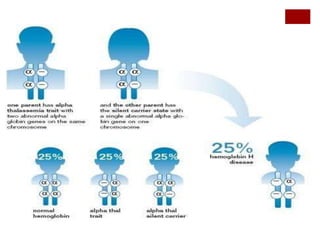
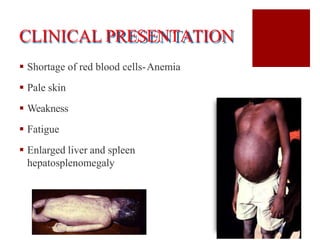








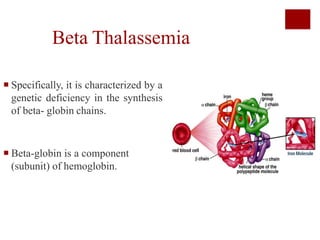


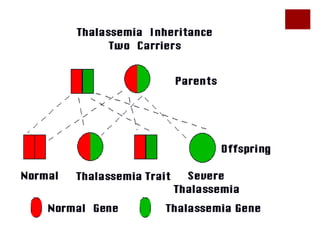

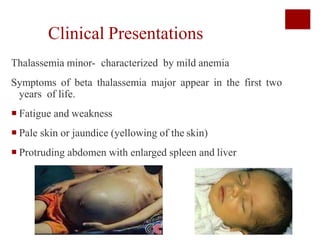









Thalassemia is an inherited blood disorder characterized by reduced or absent hemoglobin production, leading to anemia due to excessive red blood cell destruction. It is prevalent globally, particularly in India, with varying types including alpha and beta thalassemia, each demonstrating different genetic mutations and clinical presentations. Treatments typically involve blood transfusions, folate supplements, and potential bone marrow transplants, with various surgical options for managing complications.
































![THALASEMIA definition and pathophysiologyd].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/thalasemia-240515044127-8ce804c5-thumbnail.jpg?width=640&height=640&fit=bounds)






















































