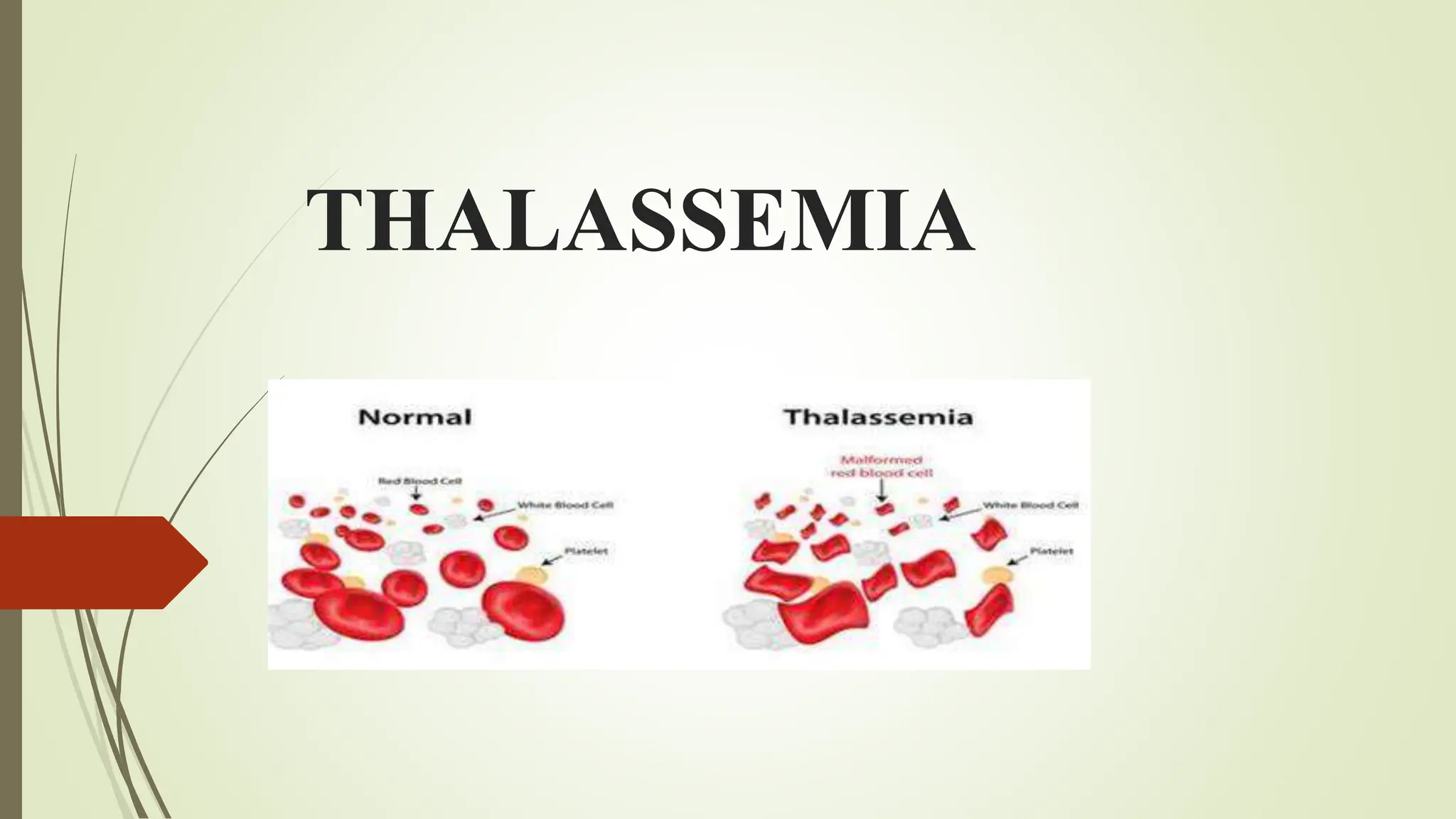
Thalassemia is an inherited blood disorder that results in reduced hemoglobin and red blood cells, leading to anemia and potential complications. There are two main types, alpha and beta thalassemia, each varying in severity based on genetic mutations affecting hemoglobin production. Diagnosis and treatment options include blood tests, transfusions, and possible bone marrow transplant, with a focus on managing symptoms and preventing complications.









































































![THALASEMIA definition and pathophysiologyd].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/thalasemia-240515044127-8ce804c5-thumbnail.jpg?width=640&height=640&fit=bounds)































![ONFH[AVN HIP] -TRIPLE REGIME -A NOVAL SURGICAL CONCEPT .pptx](https://cdn.slidesharecdn.com/ss_thumbnails/onfhavnhip2026koaconcalicutdrgokuldevdrmashraf-260210064517-213ec005-thumbnail.jpg?width=640&height=640&fit=bounds)








