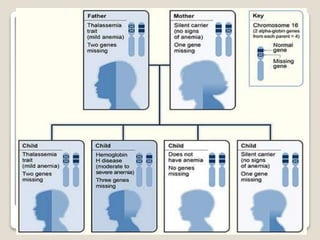
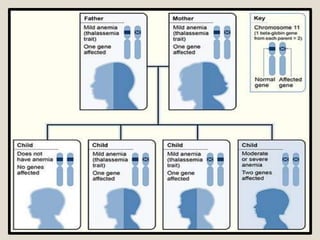
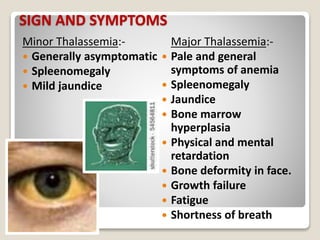
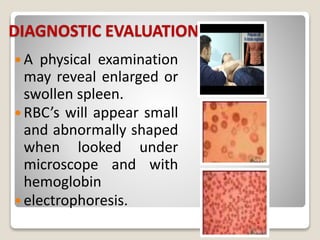
Thalassemia is a blood disorder caused by missing or mutated genes that affect hemoglobin production. It is classified based on whether the alpha or beta globin chain is affected and causes anemia. Symptoms range from mild to severe depending on the type. Diagnosis involves blood tests and genetic analysis. Treatment options include blood transfusions, iron chelation therapy, bone marrow transplants, and lifestyle management to prevent complications like infections and iron overload. Education of families on the genetic implications and lifelong management of the condition is important.