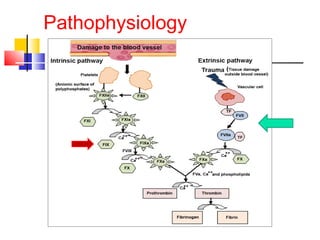
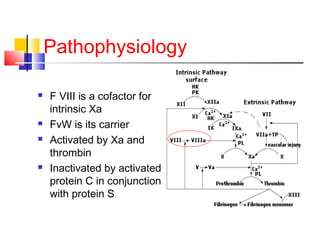
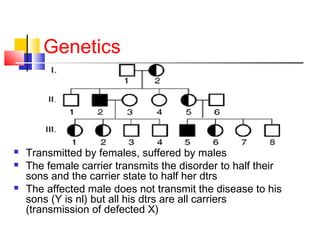
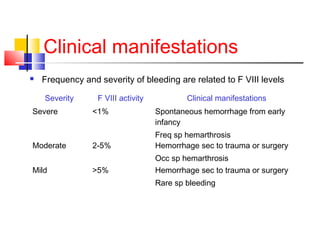
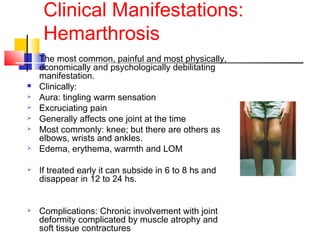
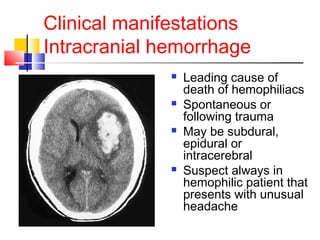
Hemophilia is a hereditary bleeding disorder caused by deficiencies in clotting factors VIII or IX. It is passed from mothers to sons and manifests as prolonged or excessive bleeding from minor injuries. The document discusses the definition, incidence, pathophysiology, genetics, clinical manifestations including hemarthrosis and treatment including factor replacement of hemophilia. It is called the "Royal Disease" because Queen Victoria was a carrier who introduced the gene into European royal families, including the Russian Tsars, affecting heirs to those thrones.