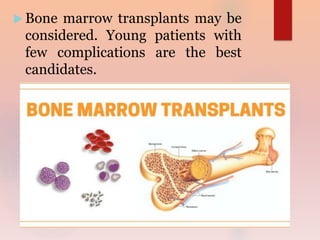
Thalassemia is a genetic blood disorder characterized by defective or reduced hemoglobin. There are two main types: alpha thalassemia affects alpha globin chain production and beta thalassemia affects beta globin chain production. Symptoms range from mild anemia to severe anemia requiring regular blood transfusions depending on which genes are affected. Treatment involves lifelong blood transfusions combined with iron chelation therapy to prevent iron overload, and potentially splenectomy or bone marrow transplant.