Downloaded 901 times
















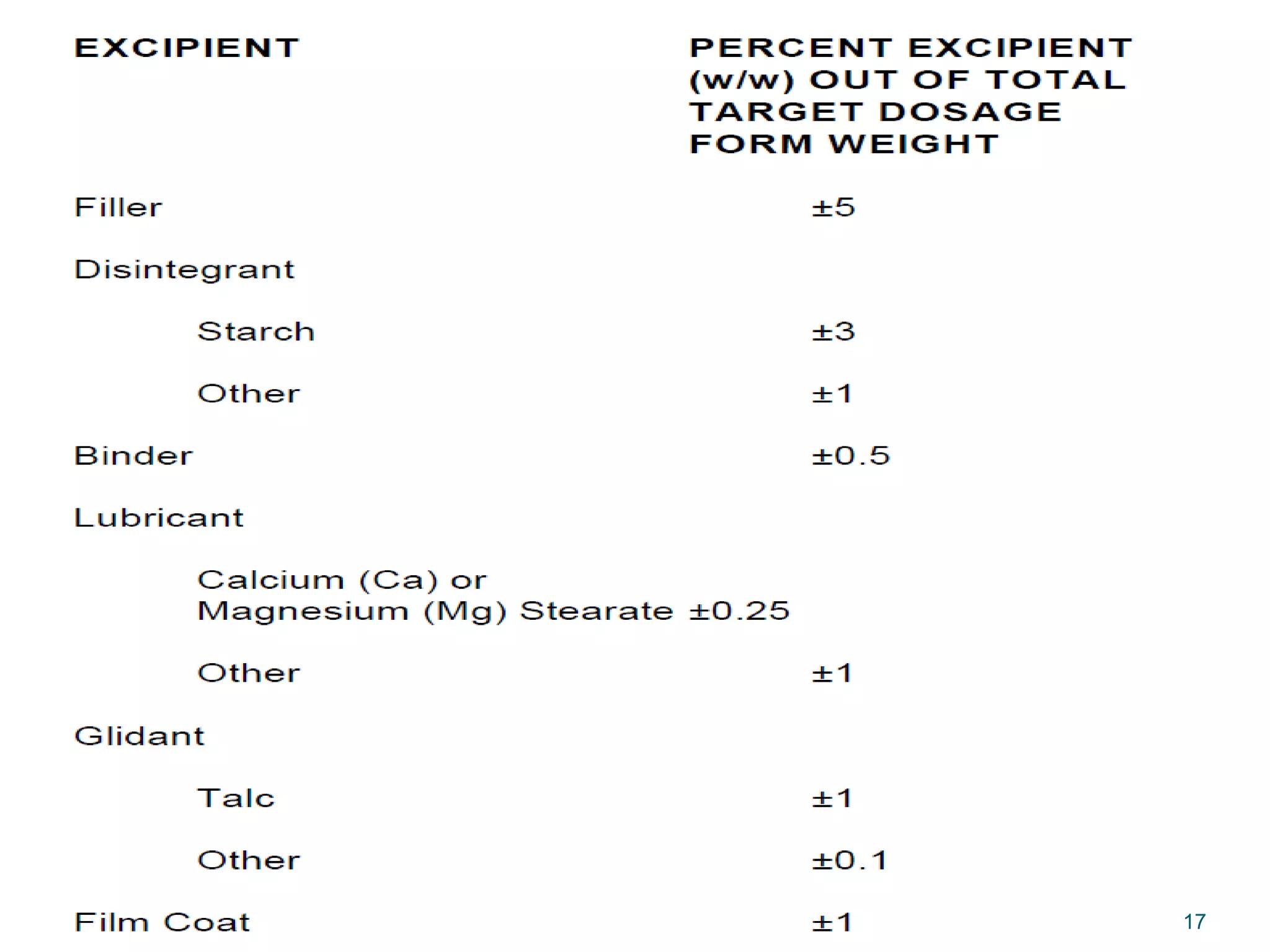



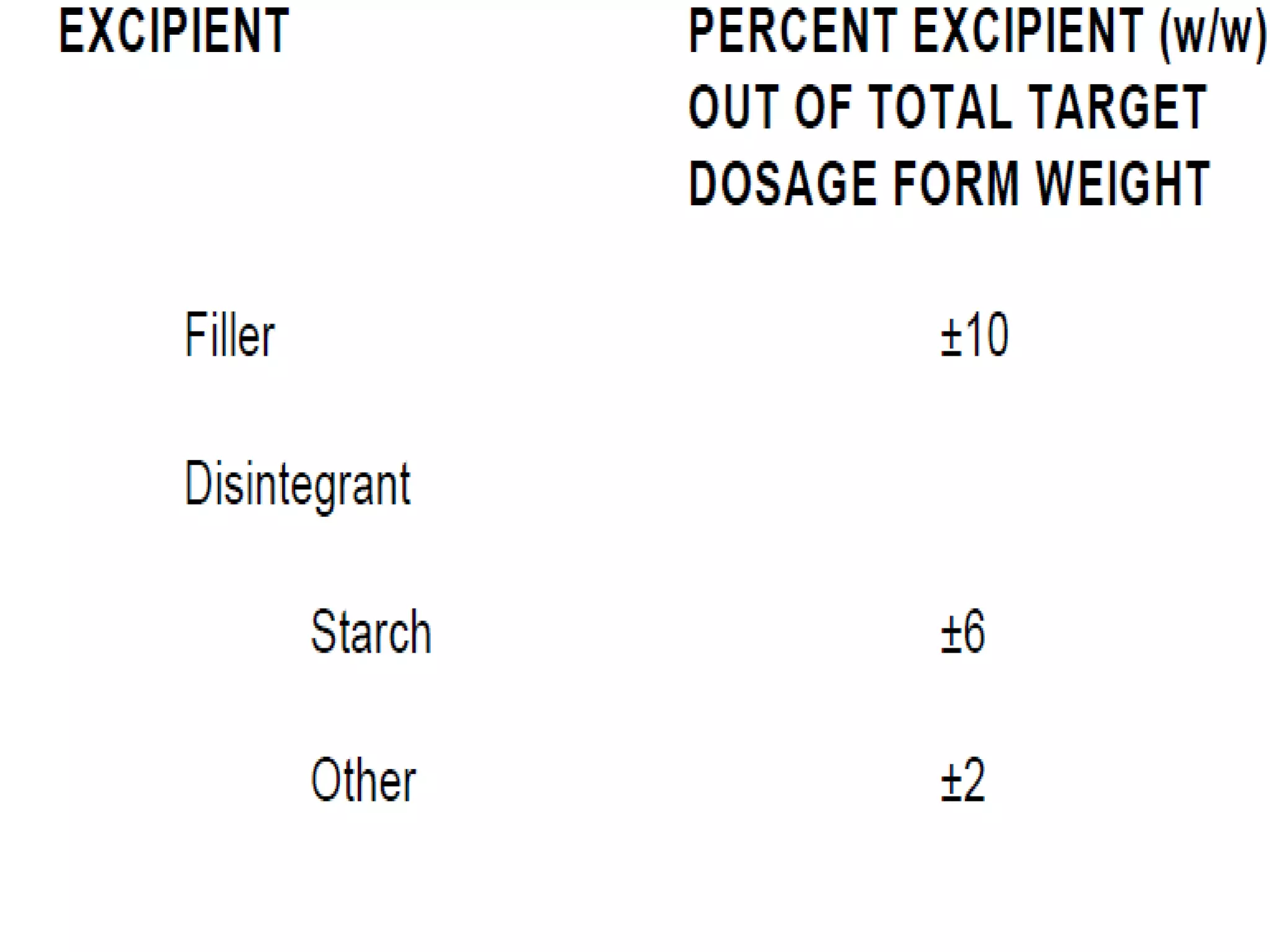
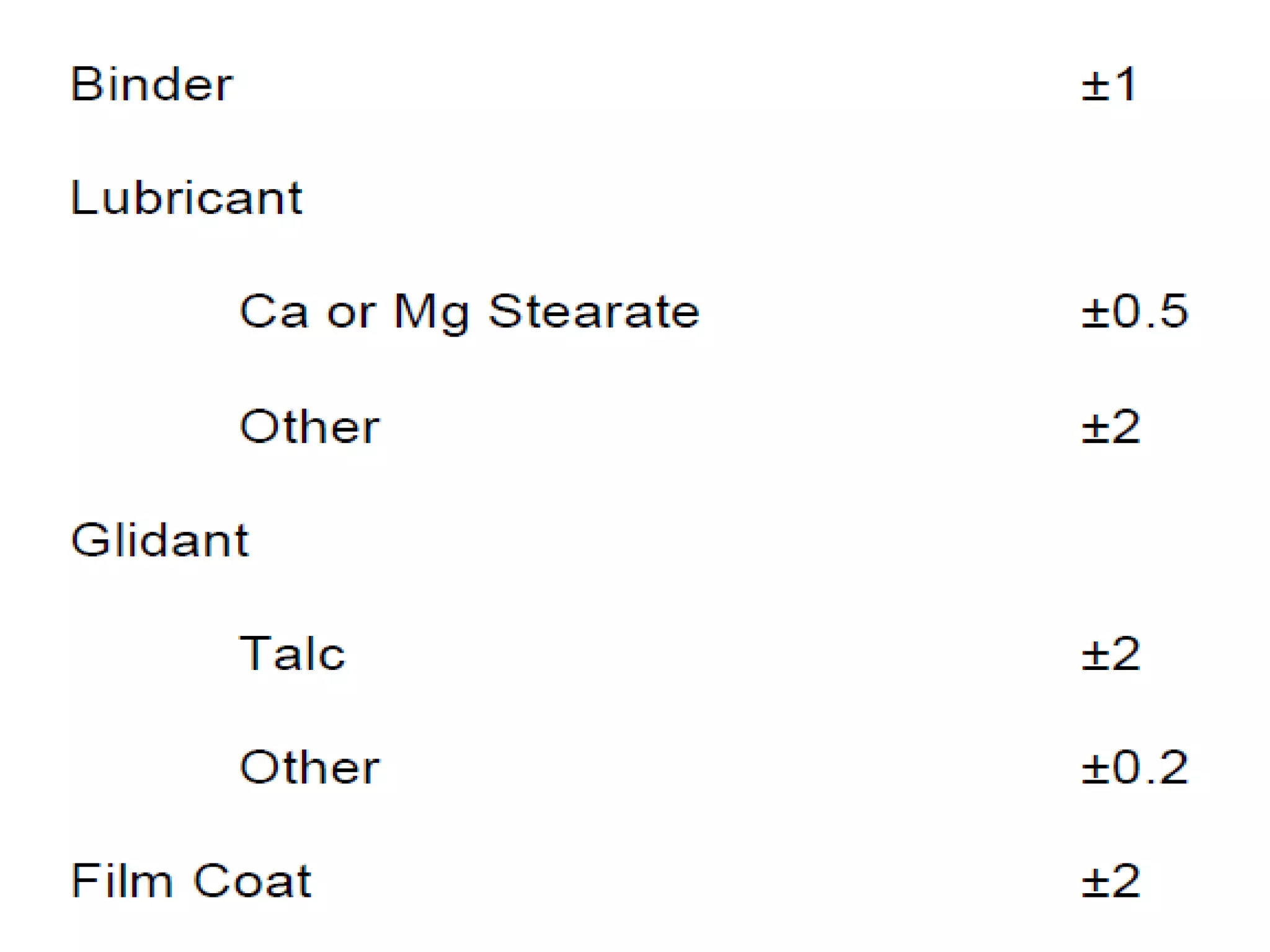













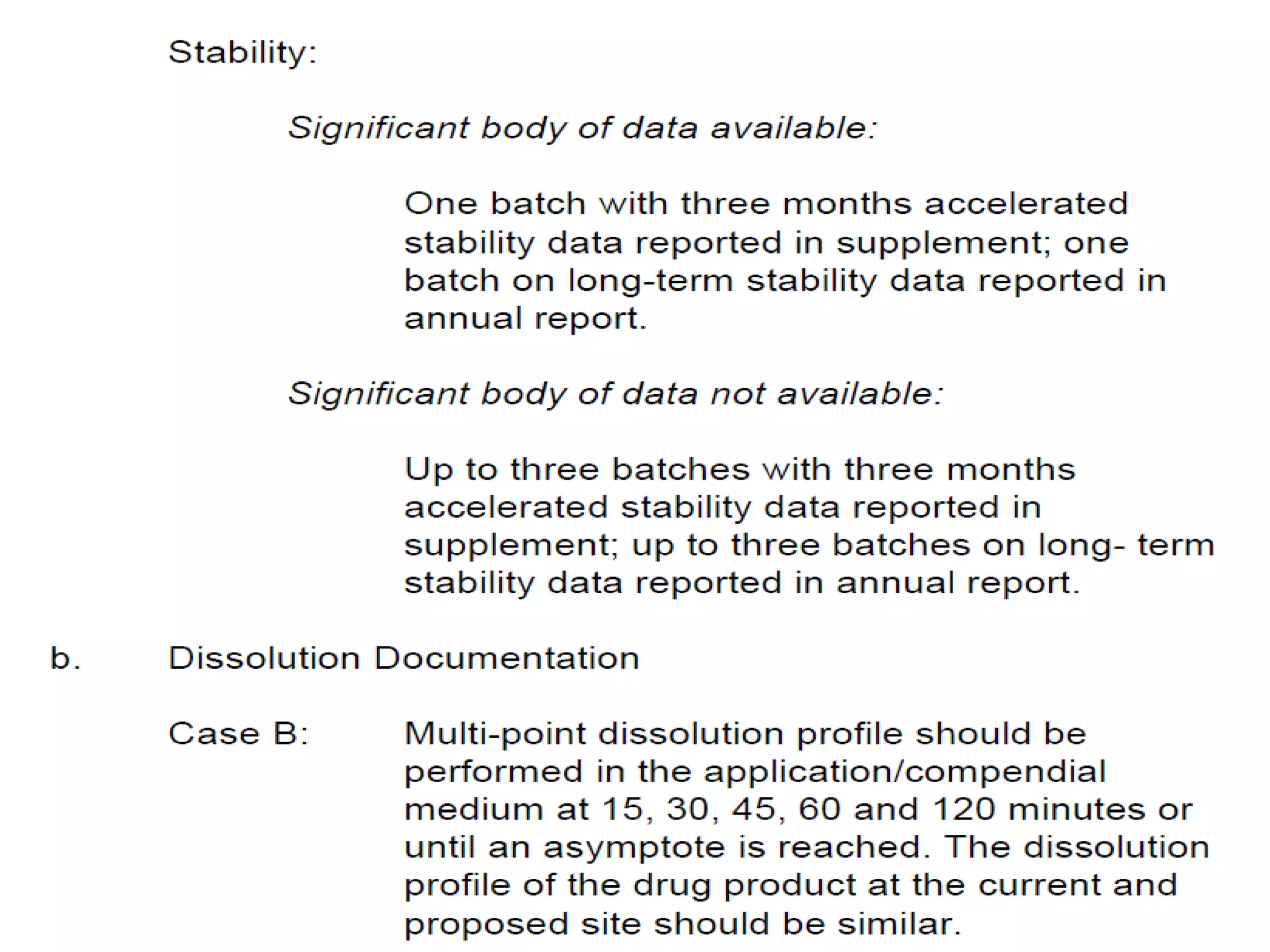

























The document discusses SUPAC (Scale-Up and Post-Approval Changes) guidances issued by the FDA for pharmaceutical manufacturing process changes. It provides an overview of existing SUPAC guidances for immediate-release solid oral dosage forms and nonsterile semisolid dosage forms. The document also describes three levels of changes - Level 1, Level 2, and Level 3 - with varying test documentation and filing requirements based on the type and extent of manufacturing changes. Guidance documents for other dosage forms are still under development.




















![cmc [ chemistry manufacturing control ]](https://cdn.slidesharecdn.com/ss_thumbnails/presentation2222ra-181120122336-thumbnail.jpg?width=640&height=640&fit=bounds)















































































