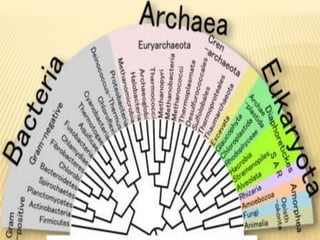
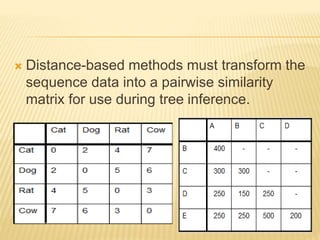
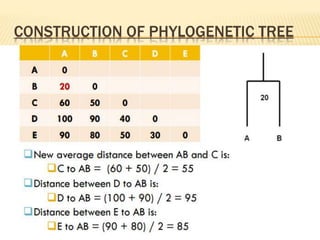
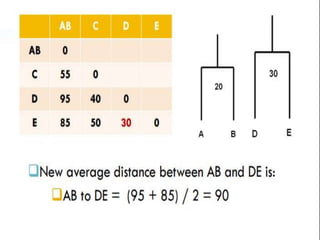
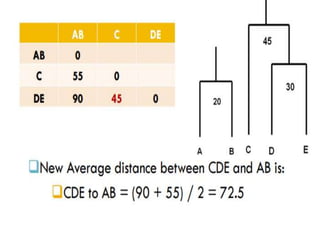
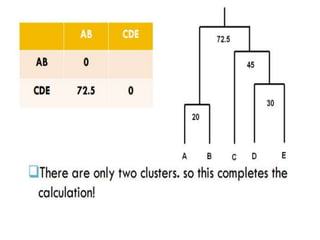
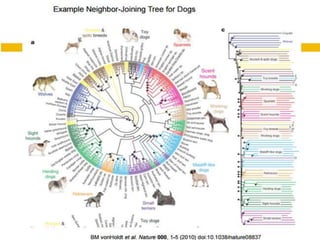
This document discusses phylogenetic tree construction using distance-based methods. It begins by introducing phylogenetic trees and their use in fields like forensics, disease prediction, and drug discovery. It then outlines the basic steps to construct a phylogenetic tree: sequence alignment, distance calculation, and tree verification. The main distance-based approaches covered are UPGMA, Neighbor-Joining, Fitch-Margoliash, Minimum Evolution. Each method calculates genetic distances differently and has advantages and limitations for reconstructing evolutionary relationships from sequence data.




















































































![Human Reproduction [ Reproductive System ] Notes @irfanullah_mehar Irfanullah...](https://cdn.slidesharecdn.com/ss_thumbnails/humanreproductionreproductivesystemnotesirfanullahmeharirfanullahmeharjanantantra-260111172350-56e85778-thumbnail.jpg?width=640&height=640&fit=bounds)


















