Downloaded 233 times



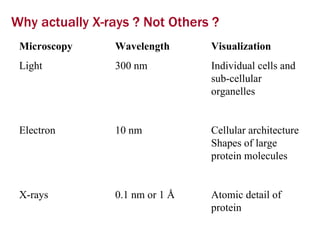

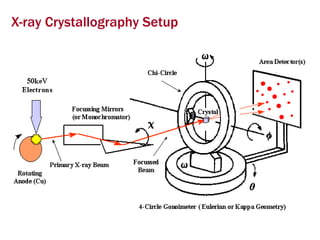
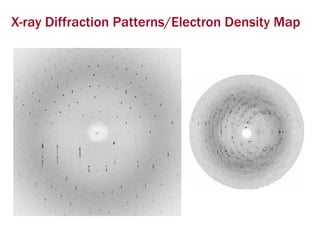

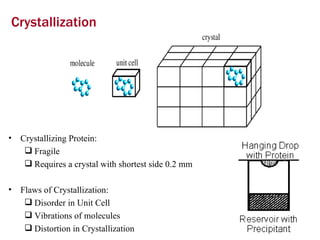





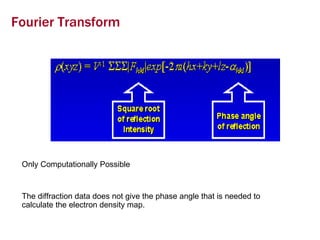
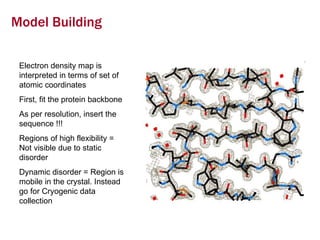




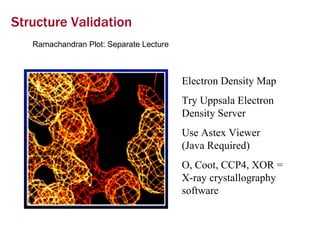


X-ray crystallography allows us to determine protein structures at the atomic level by visualizing the electron density map generated from X-ray diffraction data of protein crystals. Several steps are involved including growing high quality protein crystals, mounting crystals in the X-ray beam to collect diffraction data, solving the phase problem to produce an electron density map, building and refining an atomic model that fits the map, and validating the final protein structure. This technique provides insights into protein function and enables structure-based drug design.























































































