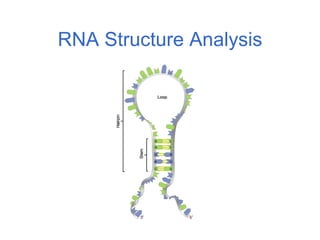
This document discusses RNA structure analysis and computational prediction of RNA secondary structure. It covers the following key points:
RNA is single-stranded but can fold into unique 3D structures guided by base pairing. There are different classes of non-coding RNAs that participate in various cellular processes. Computational prediction of RNA secondary structure is important and can be done through either ab initio or comparative approaches. Ab initio methods predict minimum free energy structures using algorithms like Nussinov and Zuker, while comparative methods analyze covariation between sequences to infer conserved structures. Common tools for RNA structure analysis include MFOLD, RNAfold and tRNAscan-SE.