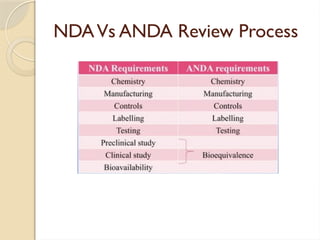
This document summarizes regulatory requirements for bioavailability and bioequivalence studies. It outlines key definitions of bioavailability and bioequivalence, requirements for studies submitted in INDs, NDAs, and ANDAs. Study designs should follow FDA guidelines and include pharmacokinetic studies using healthy volunteers. Documentation such as clinical reports and standard operating procedures must be maintained. Facilities conducting studies must be properly qualified and records retained for at least two years.