Downloaded 11 times












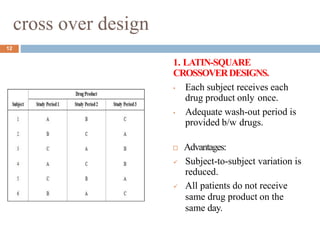
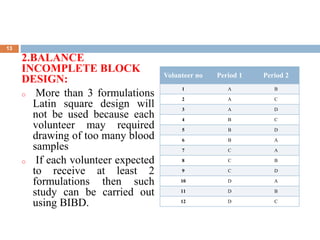

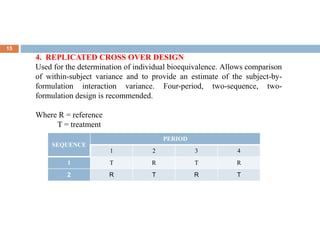










This document discusses bioequivalence studies, which compare the bioavailability of generic drugs to their branded counterparts. It covers key aspects of bioequivalence protocols such as study design, population, procedures, and data analysis. The main goals are to establish that a generic drug's absorption and exposure levels are equivalent to the reference brand name drug. Proper study design and statistical analysis are required for regulatory approval and to demonstrate therapeutic equivalence between products.











































































![ONFH[AVN HIP] -TRIPLE REGIME -A NOVAL SURGICAL CONCEPT .pptx](https://cdn.slidesharecdn.com/ss_thumbnails/onfhavnhip2026koaconcalicutdrgokuldevdrmashraf-260210064517-213ec005-thumbnail.jpg?width=640&height=640&fit=bounds)











