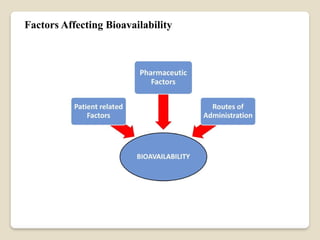
This Document Summarise about Bioavailability refers to the extent a drug is absorbed by the body. It's a critical parameter in pharmaceutical development. Formulation, route of administration, and food interactions affect bioavailability. Bioequivalence compares the bioavailability of two or more formulations.
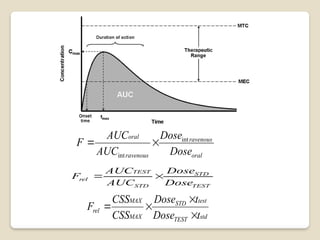
It's used to determine if a generic version is equivalent to the original. Cmax, Tmax, AUC, and half-life are key parameters for bioequivalence. Bioequivalence ensures efficacy and safety of generic versions. It's required for regulatory approval of generic drugs.Bio availability and bio equivalence are crucial in pharmaceutical development.
They ensure the quality and effectiveness of medications.