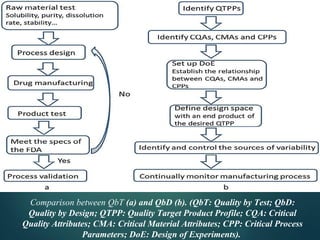
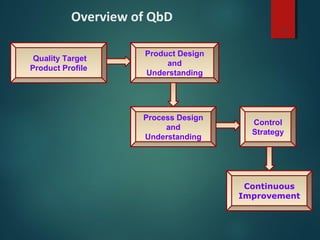
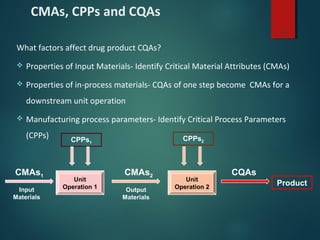
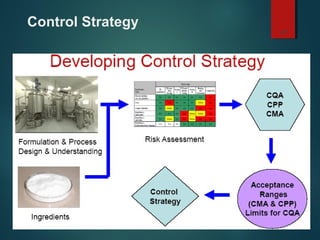
El documento aborda el concepto de 'calidad por diseño' (QbD) en el desarrollo de productos farmacéuticos, según directrices de la ICH. Se enfoca en cómo la calidad debe ser integrada en el diseño del producto en lugar de ser evaluada solamente al final, destacando la importancia de atributos críticos y estrategias de control. El QbD busca mejorar la calidad del producto, aumentar la eficiencia de desarrollo y manufactura, y reducir la variabilidad.