Download to read offline





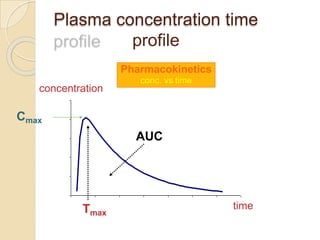

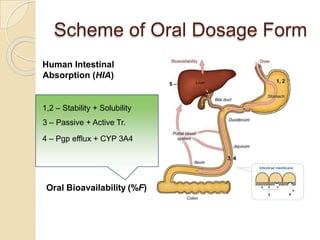



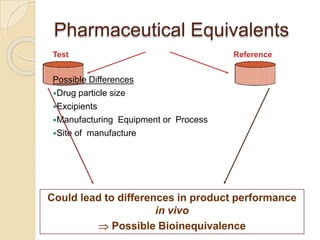





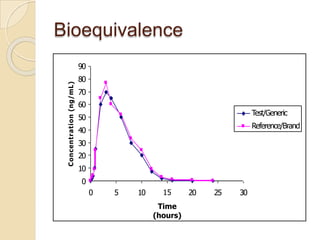

























This document discusses bioavailability and bioequivalence studies. It provides details on key pharmacokinetic parameters like AUC, Cmax, and Tmax that are evaluated in bioequivalence studies to determine if a generic drug is equivalent to a brand name drug. The document outlines current bioequivalence requirements set by various regulatory agencies like FDA, Health Canada, and others. It also discusses study design considerations, statistical analysis methods, and validation of bioanalytical methods used to evaluate bioequivalence.






































































![CASE_PRESENTATION_ON_subdural_hematoma(SDH)[1 FINAL PPT]-1.pptx](https://cdn.slidesharecdn.com/ss_thumbnails/casepresentationonsubduralhematomasdh1finalppt-1-260129172522-d405d375-thumbnail.jpg?width=640&height=640&fit=bounds)











