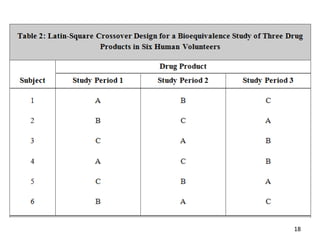
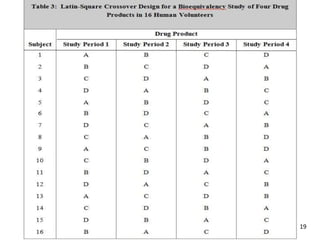
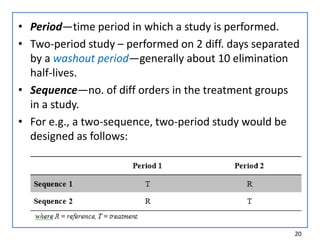
The seminar on bioequivalence studies by Muhammed Fahad focuses on the importance of comparing the bioavailability of generic drugs to brand-name products to ensure predictable drug responses. It outlines the design, necessity, and evaluation methods of bioequivalence studies, including ethical considerations and analytical methods. The document also discusses study designs like fasting studies and food intervention studies, as well as the use of waivers under specific conditions.