Protein structure prediction (1)
•Download as PPT, PDF•
10 likes•3,191 views
Protein structure prediction involves computational methods to determine a protein's 3D structure from its amino acid sequence. Ab initio methods use physics-based calculations of potential energy to predict the most stable conformation. Comparative methods leverage databases of known protein structures, searching for sequences with similar folds. Homology modeling relies on the assumption that related proteins share similar folds, allowing prediction based on matches to distant evolutionary relatives. Protein threading compares local segments of the sequence to structural fragments in databases.
Report
Share
Report
Share

Recommended
Protein Structure Prediction
The document discusses protein structure prediction. It begins by reviewing protein structure, including primary, secondary, tertiary, and quaternary structure. It then describes the building blocks of proteins, amino acids, and how their properties allow formation of regular secondary structures like alpha helices and beta sheets. The document outlines different types of secondary structure and how their patterns of hydrogen bonding influence 3D structure. It concludes by describing six classes of protein structure defined by their arrangements of alpha helices and beta sheets.
Protein fold recognition and ab_initio modeling
This document summarizes different computational methods for protein structure prediction, including homology modeling, fold recognition, threading, and ab initio modeling. Homology modeling relies on identifying proteins with similar sequences and known structures. Fold recognition and threading can be used when there are no homologs, to identify proteins with the same overall fold but different sequences. Ab initio modeling uses physics-based modeling and protein fragments to predict structure from sequence alone, and has challenges due to the vast number of possible conformations.
Protien Structure Prediction
This document discusses protein structure prediction. It begins by defining protein structure prediction as inferring a protein's three-dimensional structure from its amino acid sequence. It then outlines different levels of protein structure and some key methods for protein structure prediction, including experimental methods like X-ray crystallography and NMR, as well as computational methods like homology modeling, threading, and ab initio modeling. Specific techniques within these categories like homology modeling steps are also summarized.
Role of bioinformatics of drug designing
Bioinformatics is an interdisciplinary field that combines biology, computer science, and information technology. It enables the discovery of new biological insights and unifying principles in biology through the merging of these disciplines. There are three main sub-disciplines: developing algorithms and statistics for analyzing large datasets, analyzing various types of biological data like sequences and structures, and developing tools for accessing and managing information.
Secondary protein structure prediction
Secondary structure prediction tools analyze a protein's amino acid sequence to predict its 3D structure and function. These tools use various methods like Chou-Fasman, GOR, neural networks, and hidden Markov models to identify alpha helices and beta sheets based on characteristics like residue propensity values, sequence homology, and patterns in windows of amino acids. Accurate prediction of secondary structure is important for determining a protein's tertiary structure and biological role.
Introduction to proteomics
This document provides an introduction to proteomics presented by Shryli K S. It discusses the history of proteomics including early work using 2D gels in 1975. It describes the objectives of proteomics such as studying protein expression, function and interactions. Techniques used to study proteins are discussed including protein detection using antibodies, mass spectrometry, and protein databases. The conclusion recognizes important Nobel Prize winners that advanced the field of proteomics and proteomic applications.
Threading modeling methods
This document discusses protein threading modeling methods. Protein threading, also called fold recognition, is used to model proteins that have the same fold as proteins with known structures but no homologous sequences. It differs from homology modeling which is used for proteins that have homologous sequences. Protein threading works by using statistical knowledge of relationships between structures in the Protein Data Bank and the sequence of the protein being modeled. It is based on observations that there are a limited number of folds in nature and most new structures have similar folds to ones already in the PDB. The document then describes the general steps of the protein threading method.
Protein Threading
Protein threading is a protein structure prediction method that involves "threading" or placing an amino acid sequence into known protein structure templates to find the best matching fold. The key steps are:
1) A query sequence is threaded into structural positions of templates from a structure library to find sequence-structure alignments
2) Alignments are scored and optimized using an objective function accounting for residue interactions and preferences
3) The highest scoring template is selected as the predicted structure, though loop regions are often not accurately predicted
Recommended
Protein Structure Prediction
The document discusses protein structure prediction. It begins by reviewing protein structure, including primary, secondary, tertiary, and quaternary structure. It then describes the building blocks of proteins, amino acids, and how their properties allow formation of regular secondary structures like alpha helices and beta sheets. The document outlines different types of secondary structure and how their patterns of hydrogen bonding influence 3D structure. It concludes by describing six classes of protein structure defined by their arrangements of alpha helices and beta sheets.
Protein fold recognition and ab_initio modeling
This document summarizes different computational methods for protein structure prediction, including homology modeling, fold recognition, threading, and ab initio modeling. Homology modeling relies on identifying proteins with similar sequences and known structures. Fold recognition and threading can be used when there are no homologs, to identify proteins with the same overall fold but different sequences. Ab initio modeling uses physics-based modeling and protein fragments to predict structure from sequence alone, and has challenges due to the vast number of possible conformations.
Protien Structure Prediction
This document discusses protein structure prediction. It begins by defining protein structure prediction as inferring a protein's three-dimensional structure from its amino acid sequence. It then outlines different levels of protein structure and some key methods for protein structure prediction, including experimental methods like X-ray crystallography and NMR, as well as computational methods like homology modeling, threading, and ab initio modeling. Specific techniques within these categories like homology modeling steps are also summarized.
Role of bioinformatics of drug designing
Bioinformatics is an interdisciplinary field that combines biology, computer science, and information technology. It enables the discovery of new biological insights and unifying principles in biology through the merging of these disciplines. There are three main sub-disciplines: developing algorithms and statistics for analyzing large datasets, analyzing various types of biological data like sequences and structures, and developing tools for accessing and managing information.
Secondary protein structure prediction
Secondary structure prediction tools analyze a protein's amino acid sequence to predict its 3D structure and function. These tools use various methods like Chou-Fasman, GOR, neural networks, and hidden Markov models to identify alpha helices and beta sheets based on characteristics like residue propensity values, sequence homology, and patterns in windows of amino acids. Accurate prediction of secondary structure is important for determining a protein's tertiary structure and biological role.
Introduction to proteomics
This document provides an introduction to proteomics presented by Shryli K S. It discusses the history of proteomics including early work using 2D gels in 1975. It describes the objectives of proteomics such as studying protein expression, function and interactions. Techniques used to study proteins are discussed including protein detection using antibodies, mass spectrometry, and protein databases. The conclusion recognizes important Nobel Prize winners that advanced the field of proteomics and proteomic applications.
Threading modeling methods
This document discusses protein threading modeling methods. Protein threading, also called fold recognition, is used to model proteins that have the same fold as proteins with known structures but no homologous sequences. It differs from homology modeling which is used for proteins that have homologous sequences. Protein threading works by using statistical knowledge of relationships between structures in the Protein Data Bank and the sequence of the protein being modeled. It is based on observations that there are a limited number of folds in nature and most new structures have similar folds to ones already in the PDB. The document then describes the general steps of the protein threading method.
Protein Threading
Protein threading is a protein structure prediction method that involves "threading" or placing an amino acid sequence into known protein structure templates to find the best matching fold. The key steps are:
1) A query sequence is threaded into structural positions of templates from a structure library to find sequence-structure alignments
2) Alignments are scored and optimized using an objective function accounting for residue interactions and preferences
3) The highest scoring template is selected as the predicted structure, though loop regions are often not accurately predicted
Molecular modeling database
The Molecular Modeling Database (MMDB) is a database hosted by the National Center for Biotechnology Information that contains over 28,000 experimentally determined 3D structures of biomolecules including proteins and nucleic acids derived from the Protein Data Bank, excluding theoretical models. It facilitates computation and links structures to other data types. Each record cross-references its source PDB file. The database contains molecular structures, biological activity data, experimental data, chemical properties, and annotations to aid researchers. Examples of widely used molecular modeling databases discussed are the Protein Data Bank, PubChem, and RCSB Ligand Explorer.
Bioinformatic in drug designing
Bioinformatic tools can be applied throughout the drug design process to reduce costs and time. High-throughput screening allows testing of millions of compounds against protein targets. Computer modeling predicts compound activity and allows virtual screening. Molecular modeling visualizes compound-protein interactions to understand mechanisms of action. In silico models predict absorption, distribution, metabolism, and excretion to evaluate drug properties without animal testing. Bioinformatics databases provide protein and compound structure information to inform drug target and lead identification. Together these tools automate and accelerate key steps in drug design and development.
Homology modeling
Homology modeling is a technique used to predict the 3D structure of a protein based on the alignment of its amino acid sequence to known protein structures. It relies on the observation that structure is more conserved than sequence during evolution. The key steps in homology modeling include: 1) identifying a template structure through sequence alignment tools like BLAST, 2) correcting any errors in the initial alignment, 3) generating the protein backbone based on the template structure, 4) modeling any loops or missing regions, 5) adding side chains, 6) optimizing the model structure energetically, and 7) validating that the final model matches the template structure and has correct stereochemistry. Homology modeling is useful for applications like structure-based drug design
ProCheck
PROCheck is a tool used to evaluate protein models by analyzing residue geometry. It takes a protein structure file as input, then analyzes residue-by-residue geometry and overall structure geometry. PROCheck outputs various plots and listings that analyze features like the Ramachandran plot, residue properties, and distorted geometry regions. It aims to assess how normal or unusual a protein structure is compared to parameters from high-resolution structures.
Structural bioinformatics and pdb
Structural bioinformatics deals with prediction of 3-D structures of biological macromolecules such as proteins, DNA, RNA etc., basing on the data obtained from studies with the help of technique like X-ray crystallography, NMR etc. PDB is now essential for any study in structural biology. It is a freely accessible database of biological macromolecules.
Uni prot presentation
UniProt is a comprehensive, freely accessible database that is a central repository for protein data. It is produced through collaboration between the European Bioinformatics Institute, Swiss Institute of Bioinformatics, and Protein Information Resource. UniProt contains protein sequences, functional information, evolutionary data, and details about biological processes, post-translational modifications, interactions, and subcellular locations to characterize proteins.
Structure alignment methods
The document discusses various methods for structurally aligning proteins, including combinatorial extension, VAST, DALI, SSAP, and TM-align. It also describes Ramachandran plots, which show allowed and favored phi/psi dihedral angle combinations for protein backbone chains based on steric constraints. Structural alignment methods are useful for detecting evolutionary relationships between proteins with low sequence similarity. Ramachandran plots help validate protein structures by identifying conformations not allowed by steric hindrance.
Protein Predictinon
The document discusses experimental and computational methods for protein structure prediction. Experimental methods like NMR, X-ray crystallography, and cryo-EM can accurately determine protein structure but require isolating and crystallizing the protein. Computational methods like homology modeling, ab initio modeling, and threading/folding predict structure from sequence alone and are less accurate but do not require crystallization. Computational methods work best when a template structure is available from experimental data. While experimental methods are very accurate, they are also costly and difficult for large numbers of proteins, making computational methods a useful complement despite being less accurate.
Kegg databse
Rashi Srivastava presented on the KEGG database in biotechnology. KEGG is a database that contains genomic, chemical, and systems information to understand biological functions from the molecular level up. It includes pathways, genes, compounds, diseases, drugs, and organisms. KEGG can be searched through its flat file format using DBGET or through its relational database format for more complex queries. It also contains the KEGG MEDICUS search tool and direct SQL searches of its relational database.
Homology modelling
Just something I did for leisure :) .You need to download PDB viewer, Yasara/Accelerys for a clear understanding of this subject.
Protein 3 d structure prediction
The document discusses various computational methods for predicting the three-dimensional structure of proteins from their amino acid sequences. It describes homology modeling, which predicts structures based on known protein structural templates that share sequence homology. It also covers threading/fold recognition and ab initio modeling, which predict structures without templates by using physicochemical principles or energy minimization approaches. Key steps and programs used in each method are outlined.
methods for protein structure prediction
protein structure prediction methods. homology modelling, fold recognition, threading, ab initio methods. in short and easy form slides. after one time read you can easily understand methods for protein structure prediction.
Methods of Protein structure determination
This document summarizes several methods for determining protein structure: X-ray crystallography, nuclear magnetic resonance spectroscopy, and cryo-electron microscopy. X-ray crystallography involves growing protein crystals, exposing them to X-rays to generate diffraction patterns, and using the patterns to build 3D electron density maps of the protein. Nuclear magnetic resonance spectroscopy measures distances between atomic nuclei in soluble proteins by analyzing spectra from radiofrequency pulses applied in strong magnetic fields. Cryo-electron microscopy images frozen, hydrated protein samples with an electron microscope to determine large protein structures without the need for crystallization.
Protein Structure Determination
1) The document discusses various methods for determining the 3D structure of proteins, including x-ray crystallography, NMR spectroscopy, and cryo-electron microscopy.
2) X-ray crystallography involves purifying the protein, crystallizing it, collecting diffraction data from x-rays hitting the crystal, using this data to determine phases and calculate an electron density map, and building an atomic model through refinement.
3) NMR spectroscopy involves dissolving the purified protein and using nuclear magnetic resonance to measure distances between atomic nuclei, allowing the structure to be calculated.
HOMOLOGY MODELING IN EASIER WAY
HERE IN THIS PRESENTATION HY HOMOLOGY MODELING IS EXPLAIN , WITH EXAMPLES OF PROTEIN PRIMARY AND SECONDARY, SHOWING THE IMAGES FORM WHICH MAKES EASY TO UNDERSTAND
Kegg
The document describes several key databases within the KEGG resource, including:
- The PATHWAY database containing molecular network maps of metabolic and genetic pathways.
- The BRITE database providing hierarchical classifications of biological systems beyond what is shown in pathways.
- The LIGAND database consisting of chemical compounds, carbohydrates, reactions, and enzyme information.
KEGG aims to comprehensively capture biological knowledge through integrated databases covering genomes, pathways, diseases and drugs.
Data mining
This document provides an introduction and overview of data mining. It discusses how data mining extracts knowledge from large amounts of data to discover hidden patterns and predict future trends. It notes that for effective data mining, data sets need to be extremely large. The document outlines some key techniques of data mining including associative learning, artificial neural networks, clustering, genetic algorithms, and hidden Markov models. It also discusses applications of data mining in bioinformatics such as gene finding, protein function prediction, and disease diagnosis. Finally, it acknowledges that while bioinformatics data is rich, developing comprehensive theories remains challenging but creates opportunities for novel knowledge discovery methods.
Homology modeling of proteins (ppt)
Homology modeling, also known as comparative modeling of protein, refers to constructing an atomic-resolution model of the "target" protein from its amino acid sequence and an experimental three-dimensional structure of a related homologous protein.
Homology modeling: Modeller
Homology modeling is a computational technique for predicting the structure of a protein target based on its sequence similarity to proteins with known structures, and it involves finding a suitable template, aligning the target and template sequences, building a 3D model of the target, and evaluating the model quality. While experimental methods like X-ray crystallography and NMR can determine protein structures, they have limitations in terms of which proteins can be studied, so computational methods like homology modeling are needed to predict structures for the many proteins whose structures remain unknown.
Protein data bank
The Protein Data Bank (PDB) is an open database that archives 3D structural data of biological macromolecules. It was established in 1971 and currently holds over 150,000 structures determined by X-ray crystallography or NMR spectroscopy. The PDB is overseen by the Worldwide Protein Data Bank and freely accessible online. It serves as a key resource for structural biology and many other databases rely on protein structures deposited in the PDB.
Protein structure analysis
Proteins : is made of chain of amino acids ( amino acid= monomers) therefor the protein is polymers .
The proteins are made up of carbon, hydrogen, oxygen, and nitrogen.
Amino acid :
Computational Prediction Of Protein-1.pptx
This document discusses computational methods for predicting protein structure, including homology modeling, fold recognition/threading, and ab initio prediction. Homology modeling predicts structure based on sequence similarity to proteins with known structures. It involves aligning the target sequence to template structures, then modeling secondary structure, loops, and side chains. Accuracy depends on template quality and sequence identity above 30%. Fold recognition matches sequences to structure folds without clear homology. Ab initio prediction predicts structure from sequence alone using physics-based forces.
More Related Content
What's hot
Molecular modeling database
The Molecular Modeling Database (MMDB) is a database hosted by the National Center for Biotechnology Information that contains over 28,000 experimentally determined 3D structures of biomolecules including proteins and nucleic acids derived from the Protein Data Bank, excluding theoretical models. It facilitates computation and links structures to other data types. Each record cross-references its source PDB file. The database contains molecular structures, biological activity data, experimental data, chemical properties, and annotations to aid researchers. Examples of widely used molecular modeling databases discussed are the Protein Data Bank, PubChem, and RCSB Ligand Explorer.
Bioinformatic in drug designing
Bioinformatic tools can be applied throughout the drug design process to reduce costs and time. High-throughput screening allows testing of millions of compounds against protein targets. Computer modeling predicts compound activity and allows virtual screening. Molecular modeling visualizes compound-protein interactions to understand mechanisms of action. In silico models predict absorption, distribution, metabolism, and excretion to evaluate drug properties without animal testing. Bioinformatics databases provide protein and compound structure information to inform drug target and lead identification. Together these tools automate and accelerate key steps in drug design and development.
Homology modeling
Homology modeling is a technique used to predict the 3D structure of a protein based on the alignment of its amino acid sequence to known protein structures. It relies on the observation that structure is more conserved than sequence during evolution. The key steps in homology modeling include: 1) identifying a template structure through sequence alignment tools like BLAST, 2) correcting any errors in the initial alignment, 3) generating the protein backbone based on the template structure, 4) modeling any loops or missing regions, 5) adding side chains, 6) optimizing the model structure energetically, and 7) validating that the final model matches the template structure and has correct stereochemistry. Homology modeling is useful for applications like structure-based drug design
ProCheck
PROCheck is a tool used to evaluate protein models by analyzing residue geometry. It takes a protein structure file as input, then analyzes residue-by-residue geometry and overall structure geometry. PROCheck outputs various plots and listings that analyze features like the Ramachandran plot, residue properties, and distorted geometry regions. It aims to assess how normal or unusual a protein structure is compared to parameters from high-resolution structures.
Structural bioinformatics and pdb
Structural bioinformatics deals with prediction of 3-D structures of biological macromolecules such as proteins, DNA, RNA etc., basing on the data obtained from studies with the help of technique like X-ray crystallography, NMR etc. PDB is now essential for any study in structural biology. It is a freely accessible database of biological macromolecules.
Uni prot presentation
UniProt is a comprehensive, freely accessible database that is a central repository for protein data. It is produced through collaboration between the European Bioinformatics Institute, Swiss Institute of Bioinformatics, and Protein Information Resource. UniProt contains protein sequences, functional information, evolutionary data, and details about biological processes, post-translational modifications, interactions, and subcellular locations to characterize proteins.
Structure alignment methods
The document discusses various methods for structurally aligning proteins, including combinatorial extension, VAST, DALI, SSAP, and TM-align. It also describes Ramachandran plots, which show allowed and favored phi/psi dihedral angle combinations for protein backbone chains based on steric constraints. Structural alignment methods are useful for detecting evolutionary relationships between proteins with low sequence similarity. Ramachandran plots help validate protein structures by identifying conformations not allowed by steric hindrance.
Protein Predictinon
The document discusses experimental and computational methods for protein structure prediction. Experimental methods like NMR, X-ray crystallography, and cryo-EM can accurately determine protein structure but require isolating and crystallizing the protein. Computational methods like homology modeling, ab initio modeling, and threading/folding predict structure from sequence alone and are less accurate but do not require crystallization. Computational methods work best when a template structure is available from experimental data. While experimental methods are very accurate, they are also costly and difficult for large numbers of proteins, making computational methods a useful complement despite being less accurate.
Kegg databse
Rashi Srivastava presented on the KEGG database in biotechnology. KEGG is a database that contains genomic, chemical, and systems information to understand biological functions from the molecular level up. It includes pathways, genes, compounds, diseases, drugs, and organisms. KEGG can be searched through its flat file format using DBGET or through its relational database format for more complex queries. It also contains the KEGG MEDICUS search tool and direct SQL searches of its relational database.
Homology modelling
Just something I did for leisure :) .You need to download PDB viewer, Yasara/Accelerys for a clear understanding of this subject.
Protein 3 d structure prediction
The document discusses various computational methods for predicting the three-dimensional structure of proteins from their amino acid sequences. It describes homology modeling, which predicts structures based on known protein structural templates that share sequence homology. It also covers threading/fold recognition and ab initio modeling, which predict structures without templates by using physicochemical principles or energy minimization approaches. Key steps and programs used in each method are outlined.
methods for protein structure prediction
protein structure prediction methods. homology modelling, fold recognition, threading, ab initio methods. in short and easy form slides. after one time read you can easily understand methods for protein structure prediction.
Methods of Protein structure determination
This document summarizes several methods for determining protein structure: X-ray crystallography, nuclear magnetic resonance spectroscopy, and cryo-electron microscopy. X-ray crystallography involves growing protein crystals, exposing them to X-rays to generate diffraction patterns, and using the patterns to build 3D electron density maps of the protein. Nuclear magnetic resonance spectroscopy measures distances between atomic nuclei in soluble proteins by analyzing spectra from radiofrequency pulses applied in strong magnetic fields. Cryo-electron microscopy images frozen, hydrated protein samples with an electron microscope to determine large protein structures without the need for crystallization.
Protein Structure Determination
1) The document discusses various methods for determining the 3D structure of proteins, including x-ray crystallography, NMR spectroscopy, and cryo-electron microscopy.
2) X-ray crystallography involves purifying the protein, crystallizing it, collecting diffraction data from x-rays hitting the crystal, using this data to determine phases and calculate an electron density map, and building an atomic model through refinement.
3) NMR spectroscopy involves dissolving the purified protein and using nuclear magnetic resonance to measure distances between atomic nuclei, allowing the structure to be calculated.
HOMOLOGY MODELING IN EASIER WAY
HERE IN THIS PRESENTATION HY HOMOLOGY MODELING IS EXPLAIN , WITH EXAMPLES OF PROTEIN PRIMARY AND SECONDARY, SHOWING THE IMAGES FORM WHICH MAKES EASY TO UNDERSTAND
Kegg
The document describes several key databases within the KEGG resource, including:
- The PATHWAY database containing molecular network maps of metabolic and genetic pathways.
- The BRITE database providing hierarchical classifications of biological systems beyond what is shown in pathways.
- The LIGAND database consisting of chemical compounds, carbohydrates, reactions, and enzyme information.
KEGG aims to comprehensively capture biological knowledge through integrated databases covering genomes, pathways, diseases and drugs.
Data mining
This document provides an introduction and overview of data mining. It discusses how data mining extracts knowledge from large amounts of data to discover hidden patterns and predict future trends. It notes that for effective data mining, data sets need to be extremely large. The document outlines some key techniques of data mining including associative learning, artificial neural networks, clustering, genetic algorithms, and hidden Markov models. It also discusses applications of data mining in bioinformatics such as gene finding, protein function prediction, and disease diagnosis. Finally, it acknowledges that while bioinformatics data is rich, developing comprehensive theories remains challenging but creates opportunities for novel knowledge discovery methods.
Homology modeling of proteins (ppt)
Homology modeling, also known as comparative modeling of protein, refers to constructing an atomic-resolution model of the "target" protein from its amino acid sequence and an experimental three-dimensional structure of a related homologous protein.
Homology modeling: Modeller
Homology modeling is a computational technique for predicting the structure of a protein target based on its sequence similarity to proteins with known structures, and it involves finding a suitable template, aligning the target and template sequences, building a 3D model of the target, and evaluating the model quality. While experimental methods like X-ray crystallography and NMR can determine protein structures, they have limitations in terms of which proteins can be studied, so computational methods like homology modeling are needed to predict structures for the many proteins whose structures remain unknown.
Protein data bank
The Protein Data Bank (PDB) is an open database that archives 3D structural data of biological macromolecules. It was established in 1971 and currently holds over 150,000 structures determined by X-ray crystallography or NMR spectroscopy. The PDB is overseen by the Worldwide Protein Data Bank and freely accessible online. It serves as a key resource for structural biology and many other databases rely on protein structures deposited in the PDB.
What's hot (20)
Similar to Protein structure prediction (1)
Protein structure analysis
Proteins : is made of chain of amino acids ( amino acid= monomers) therefor the protein is polymers .
The proteins are made up of carbon, hydrogen, oxygen, and nitrogen.
Amino acid :
Computational Prediction Of Protein-1.pptx
This document discusses computational methods for predicting protein structure, including homology modeling, fold recognition/threading, and ab initio prediction. Homology modeling predicts structure based on sequence similarity to proteins with known structures. It involves aligning the target sequence to template structures, then modeling secondary structure, loops, and side chains. Accuracy depends on template quality and sequence identity above 30%. Fold recognition matches sequences to structure folds without clear homology. Ab initio prediction predicts structure from sequence alone using physics-based forces.
Molecular modelling (1)
This document presents an overview of molecular modeling techniques. It discusses the history of molecular modeling and some common computational methods like molecular mechanics, quantum mechanics and molecular dynamics. It also describes different modeling approaches like template modeling techniques such as homology modeling and threading as well as template-free modeling methods including ab initio and knowledge-based modeling. The document concludes that molecular modeling can provide useful insights for research if used carefully while also noting current limitations, especially for modeling larger protein structures.
Computer Aided Molecular Modeling
This document discusses protein structure prediction and molecular modeling. It begins with an overview of the druggable genome and protein structure prediction approaches such as ab initio modeling, threading, and homology modeling. It then provides details on homology modeling steps including searching databases, selecting templates, aligning sequences, building models, and model evaluation. The document also discusses protein-ligand docking, scoring functions, assessing docking performance, and practical aspects of docking such as protein and ligand preparation.
Structural bioinformatics.
This document provides an overview of structural bioinformatics and protein structure. It discusses the basics of protein structure including the primary, secondary, tertiary, and quaternary levels. Methods for determining three-dimensional protein structure such as X-ray crystallography and NMR spectroscopy are described. The document also covers protein structure visualization tools, comparison of protein structures to determine evolutionary relationships, and classification databases like SCOP and CATH. The key applications of structural bioinformatics are structure-based function prediction and aiding structural genomics projects.
threading and homology modelling methods
The document discusses protein structure prediction methods such as homology modeling and threading. Homology modeling relies on sequence similarity between the target and template proteins to generate a structural model. It involves aligning the sequences, building the backbone based on the template, and modeling side chains. Threading methods can be used when sequence similarity is low but still detects structural similarity by identifying conserved protein folds from structural databases. Experimental techniques like X-ray crystallography and NMR spectroscopy determine protein structures but have limitations for some proteins.
In silico structure prediction
(1) There are four levels of protein structure: primary, secondary, tertiary, and quaternary. Experimental methods like X-ray crystallography and NMR spectroscopy can determine protein structures but are expensive and time-consuming. (2) Computational structure prediction methods include homology/comparative modeling, protein threading, and ab initio modeling. Homology modeling is most reliable when the sequence identity is over 30-50% to a template with a known structure. (3) Protein threading is used when there is no clear homolog but the protein may have the same fold as one in PDB. It aligns sequences to structures and evaluates fitness to predict the model.
Computational Prediction of Protein Structure.pptx
This presentation will give you information about Computational Prediction of Protein Structure and will cover Threading and Homology Modelling.
protein Modeling Abi.pptx
This document discusses different methods for predicting the secondary structure of proteins, including statistical methods like Chou-Fasman and GOR that use amino acid frequencies, and neural network methods like PHD that use multiple sequence alignments and training sets of known structures. It also briefly outlines experimental methods for determining protein structure like X-ray crystallography, NMR spectroscopy, and cryo-electron microscopy.
Motif & Domain
This document discusses motifs and domains in proteins. It defines motifs as short conserved regions related to function, such as binding sites, that are not detectable by sequence searches. There are sequence motifs consisting of nucleotide or amino acid patterns, and structural motifs formed by amino acid spatial arrangements. Domains are stable, independently folding units of proteins that determine structure and function. Both motifs and domains are useful for classifying protein families and have structural and functional roles, though domains are more stable independently. Motifs and domains form through interactions of alpha helices and beta sheets and have similarities, but domains mainly determine unique functions while motifs mainly provide structural roles within families.
Protein 3D structure and classification database
This document discusses various aspects of protein structure and modeling techniques. It begins with an introduction to proteins and their basic structures. It then discusses the primary structures of proteins including amino acids. Later, it describes different levels of protein structure such as secondary structure involving alpha helices and beta sheets, tertiary structure involving the overall shape of the protein, and quaternary structure involving multiple polypeptide chains. The document also discusses modeling techniques like threading/fold recognition to predict structure based on sequence similarity and ab initio modeling to predict structure from sequence alone.
L1Protein_Structure_Analysis.pptx
The document discusses protein structure modeling through homology modeling. It describes the key steps in homology modeling which include: (1) finding a suitable template through database searches, (2) aligning the target sequence to the template, (3) assigning coordinates from conserved regions of the template, (4) building loops and variable regions either from other structures or de novo, (5) searching for optimal side chain conformations, and (6) refining the model through molecular mechanics. The document emphasizes validating the final model to identify any inherent errors from the template or modeling process.
Intro to in silico drug discovery 2014
This document provides an overview of in silico drug design with a focus on safety and efficacy. It discusses using computational approaches to model molecular interactions for small molecule drugs and biologics. For small molecules, it describes obtaining protein structures, simulating binding via docking, and considering absorption and toxicity. For biologics like antibodies, it discusses engineering for reduced immunogenicity and improved half-life via Fc modifications. The goal is developing safer, more effective therapies through computational analysis and protein design.
De novo str_prediction
This document discusses de novo protein structure prediction, which predicts protein structure from amino acid sequence alone without using existing protein templates. It notes the need for ab initio prediction when no homologous structures exist. Successful de novo prediction requires an accurate energy function to identify native structures, an efficient conformational search method, and ability to select native models. Results from ab initio prediction typically have 5-10 Angstrom accuracy. Domain prediction is important to divide large proteins into independently folding domains for prediction. Advantages include automation and ability to structurally annotate genomes. Challenges include the vast conformational search space and need for accurate energy functions.
Bioinformatica t7-protein structure
The document discusses various topics in bioinformatics and protein structure. It provides an overview of ongoing thesis topics at Biobix including biomarker prediction, methylation, metabolomics, peptidomics, and more. It also discusses the rationale for understanding protein structure and function, levels of protein structure from primary to quaternary, methods for determining structure like X-ray crystallography, and approaches to secondary structure prediction including Chou-Fasman.
protein design, principles and examples.pptx
Protein design uses structural biology knowledge to predict amino acid sequences that produce proteins with targeted properties, allowing hypotheses to be tested. Computational methods now design proteins very different from known ones. Protein design remains an important problem, and current methods largely use physics-based approaches relying on single structures, despite multiple structures being available. A new method called FlexiBaL-GP uses machine learning to learn lower dimensional representations of backbone movements from multiple structures.
Nanorobotic Devices Of Nature
The following presentation is only for quick reference. I would advise you to read the theoretical aspects of the respective topic and then use this presentation for your last minute revision. I hope it helps you..!!
Mayur D. Chauhan
Presentation1
Homology modeling is a computational method to predict the 3D structure of a protein based on the known structure of homologous proteins. It involves 7 main steps: 1) selecting a template protein with high sequence similarity, 2) aligning the sequences, 3) building the protein backbone, 4) modeling loops and insertions/deletions, 5) refining side chains, 6) refining the overall structure using energy minimization, and 7) evaluating the model. Homology models can accurately predict protein structure when the sequence identity between the target and template is above 30%. Models are useful for studying protein function and designing drugs.
structure of proteins
This document discusses the structure of proteins at four levels: primary, secondary, tertiary, and quaternary. The primary structure refers to the amino acid sequence in the polypeptide chain. Secondary structures include alpha helices and beta sheets formed by hydrogen bonding between amino acids. Tertiary structure describes the overall 3D shape of the protein formed by interactions between amino acid side chains. Quaternary structure applies to proteins composed of multiple polypeptide subunits that combine through non-covalent bonds. The structures are determined through techniques like X-ray crystallography and NMR spectroscopy.
structure of protins
This document discusses the structure of proteins at four levels: primary, secondary, tertiary, and quaternary. The primary structure refers to the amino acid sequence in the polypeptide chain. Secondary structures include alpha helices and beta sheets formed by hydrogen bonding between amino acids. Tertiary structure describes the overall 3D shape of the protein formed by interactions between amino acid side chains. Quaternary structure applies to proteins composed of multiple polypeptide subunits that combine through non-covalent bonds. The structures are determined through techniques like X-ray crystallography and NMR spectroscopy.
Similar to Protein structure prediction (1) (20)
Computational Prediction of Protein Structure.pptx
Computational Prediction of Protein Structure.pptx
More from Sabahat Ali
RECOMBINATION MOLECULAR BIOLOGY PPT UPDATED new.pptx
This ppt is about recombination and where it occurs. Types of recombination and models of recombination along with many factors in prokaryotic and eukaryotic recombination
Good laboratory practices in a pharmaceutical lab 1
This document discusses good laboratory practices in a pharmaceutical lab. It outlines the members of a group project on this topic and provides an introduction to pharmaceutical lab testing. It then covers topics like GMP, GLP, quality control, quality assurance, reducing human errors, and the scope of QA and QC in a pharmaceutical lab. Key points include that pharmaceutical labs test raw materials, finished products, and conduct validation, stability, and analytical method development testing. GMP and GLP aim to minimize risks and ensure consistent quality production. QA and QC work to guarantee drug quality and safety at all stages from development to sales.
Degradation of PLA at Mesophillic and thermophillic conditions
This document summarizes research on the degradation of polylactic acid (PLA) under mesophilic and thermophilic conditions. Key findings include:
1) Mesophilic bacteria like Pseudomonas geniculata and Streptomyces pavanii were found to degrade PLA films at 25-40°C, with S. pavanii showing higher degradation.
2) PLA degradation was higher under thermophilic (41-122°C) conditions compared to mesophilic (20-45°C) due to PLA-degrading enzymes working best at high temperatures. Up to 90% of PLA weight loss was observed at thermophilic temperatures within 12 days of
Life cycle Assesment and waste stratigies of PLA
Group 2 presented on strategies for polylactic acid (PLA) waste, including recycling and biodegradation. There are three main routes for producing PLA: polymerization of lactic acid monomers, condensation of lactic acid, and fermentation. PLA can be chemically recycled through hydrolytic or alcoholytic depolymerization. An innovative process called the Zeus Waste PLA Depolymerization Process uses solvents like chloroform and alcohols like methanol at low temperatures to break PLA down into its original lactic acid monomers. PLA biodegrades through hydrolysis of ester bonds, thermal degradation, and photodegradation when exposed to sunlight.
Environmental biodegradation of PLA by Biotic and Abiotic factors
PLA is a biodegradable polymer that can degrade through both biotic and abiotic factors in the environment. Biotic degradation occurs through the action of microorganisms like bacteria and fungi that produce enzymes to break down PLA. Specific bacteria identified to degrade PLA include species of Pseudomonas and Streptomyces. Fungal degradation is also possible, with Phanerochaete chrysosporium shown to effectively degrade PLA. Abiotic degradation happens through hydrolysis when water breaks the ester bonds of PLA, which is accelerated at higher temperatures and pH levels.
Energy expenditure and BMR
The document discusses energy expenditure and basal metabolic rate (BMR). It defines energy expenditure as the amount of energy needed for bodily functions like breathing and circulation, while BMR is the minimum energy required for essential physiological processes when at rest. The document outlines several factors that affect BMR, such as age, gender, weight, and thyroid function. Maintaining caloric balance between intake and expenditure through diet and exercise can prevent weight gain.
Agriculture applications of nanobiotechnology
This document discusses the potential applications of nanobiotechnology in agriculture. It begins by introducing how nanoparticles can interact with agricultural hosts and tissues. It then discusses several specific applications, including using nanoparticles for plant disease management and diagnostics, as well as for delivering pesticides, nutrients, and plant hormones. The document also notes potential applications in areas like recycling agricultural waste, soil improvement, water purification, and plant breeding. It acknowledges both the promise and challenges of nanotechnology for modernizing agriculture to address issues like increasing food supply to support population growth amid changing environmental conditions.
Macronutrients and nutrition
Macronutrients provide energy and are essential for growth and maintenance of the body. The document discusses the three main macronutrients - carbohydrates, proteins, and fats. Carbohydrates are divided into simple and complex categories, with simple carbs like sugars providing quick energy and complex carbs like whole grains being more filling and nutritious. Proteins are essential building blocks and energy sources, with animal products providing complete proteins and plant sources providing complementary proteins when combined. Fats serve various functions in the body and are classified based on their structure.
Poly lactic Acid Biodegradation
The document discusses methods to enhance the biodegradation of polylactic acid (PLA). It analyzes modifications to PLA's physical properties and amending the environment with various factors like stimulants. It summarizes that biodegradation of PLA mainly occurs through hydrolysis of ester bonds and is induced by microorganisms like certain actinomycetes, bacteria, and fungi. Key factors like temperature, pH, humidity, and oxygen levels also affect the degradation rate. While PLA is biodegradable, the process is often slow under natural conditions.
Alzhemier's disease and koraskoff syndrome
Alzheimer's disease, Korsakoff's syndrome, and dreaming are compared and contrasted. Alzheimer's disease results from neuronal death and synapse loss, causing memory loss and dementia. Korsakoff's syndrome is caused by thiamine deficiency and can be reversed if treated early. Dreams occur during REM sleep and may help with memory consolidation. Both diseases involve memory loss and neuronal/synaptic changes, while dreaming is a normal process that occurs during sleep and differs in its effects on memory and brain activity.
Nerve cells, Nervous communication & its link to the celllular signalling
The document discusses the structure and function of neurons. It notes that neurons are specialized cells that communicate via electrical and chemical signals. They contain dendrites that receive signals, a cell body, and an axon that transmits signals. At synapses, chemical neurotransmitters transmit signals between neurons or to other cell types. Neurons form circuits that allow for complex coordinated responses. The action potential involves changes in ion channel permeability that propagate electrical signals rapidly along axons. Calcium acts as an important intracellular messenger in neurons and other cell types, often working through the calcium sensor protein calmodulin.
Peptide Hormones and Catecholamines
Peptide hormones and catecholamines allow for rapid responses to environmental changes. They are stored in secretory vesicles and released via exocytosis within seconds or minutes in response to stimulation. This causes short-term effects that are terminated once the hormones are degraded. In contrast, steroid hormones and thyroid hormones are synthesized from cholesterol or thyroglobulin precursors within cells. They diffuse out of cells and circulate in the blood bound to carrier proteins. This allows their effects to last longer, from hours to days, but production and release takes longer than for peptide hormones and catecholamines. The different hormone types thus allow for both rapid short-term responses and longer-term regulatory effects.
Membrane Proteins & its types
Integral & Peripheral membrane proteins
Helical membrane proteins could be single pass /multi pass
porins which are beta sheets ion channels.
membrane lipids & its types
lipids are categorized into 4 major categories.
1) phospholipids
2) glycolipids
3) steroids
4) archeal membrane lipids
Biomembranes (lipids, proteins, carbohydrates)
It includes composition of membranes (fluid mosaic model)
where these membranes are present?
what functions membranes perform?
cell to cell signalling
Cells in multicellular organisms communicate through elaborate signaling networks involving hundreds of signaling molecules. These molecules allow cells to regulate development, growth, and coordinated function. Signaling occurs through paracrine, synaptic, and endocrine mechanisms using molecules like hormones, neurotransmitters, and growth factors. Target cells contain receptors that recognize signaling molecules with high specificity and affinity. While some responses are rapid, others involve long-term changes through regulated synthesis, release, and degradation of signaling compounds.
Protein Folding Mechanism
Folding depends upon sequence of Amino Acids not the Composition. Folding starts with the secondary structure and ends at quaternary structure.
Denaturation occur at secondary, tertiary & quaternary level but not at primary level.
Proetin Tertiary Structure
Tertiary Structure basically of Hydrophobic interactions, (interactions in side chains), hydrogen bonding, salt bridges, Vander Waals interactions.
e.g. Globular proteins & Fibrous Proteins
Restriction digestion
Restriction Enzymes are used to digest DNA at specific Restriction sites(flanking ends) to produce ends that are either cohesive or blunt.
Polymerase Chain Reaction(PCR)
Amplifying DNA through polymerase chain reaction,
3 steps in PCR
Denaturation
Annealing
Extension
More from Sabahat Ali (20)
RECOMBINATION MOLECULAR BIOLOGY PPT UPDATED new.pptx
RECOMBINATION MOLECULAR BIOLOGY PPT UPDATED new.pptx
Good laboratory practices in a pharmaceutical lab 1
Good laboratory practices in a pharmaceutical lab 1
Degradation of PLA at Mesophillic and thermophillic conditions
Degradation of PLA at Mesophillic and thermophillic conditions
Environmental biodegradation of PLA by Biotic and Abiotic factors
Environmental biodegradation of PLA by Biotic and Abiotic factors
Nerve cells, Nervous communication & its link to the celllular signalling
Nerve cells, Nervous communication & its link to the celllular signalling
Recently uploaded
Summary Of transcription and Translation.pdf
Hello Everyone Here We are Sharing You with The process of protien Synthesis in very short points you will be Able to understand It Very well
Pests of Storage_Identification_Dr.UPR.pdf
InIndia-post-harvestlosses-unscientificstorage,insects,rodents,micro-organismsetc.,accountforabout10percentoftotalfoodgrains
Graininfestation
Directdamage
Indirectly
•theexuviae,skin,deadinsects
•theirexcretawhichmakefoodunfitforhumanconsumption
About600speciesofinsectshavebeenassociatedwithstoredgrainproducts
100speciesofinsectpestsofstoredproductscauseeconomiclosses
Randomised Optimisation Algorithms in DAPHNE
Slides from talk:
Aleš Zamuda: Randomised Optimisation Algorithms in DAPHNE .
Austrian-Slovenian HPC Meeting 2024 – ASHPC24, Seeblickhotel Grundlsee in Austria, 10–13 June 2024
https://ashpc.eu/
Mending Clothing to Support Sustainable Fashion_CIMaR 2024.pdf
Ozturkcan, S., Berndt, A., & Angelakis, A. (2024). Mending clothing to support sustainable fashion. Presented at the 31st Annual Conference by the Consortium for International Marketing Research (CIMaR), 10-13 Jun 2024, University of Gävle, Sweden.
MICROBIAL INTERACTION PPT/ MICROBIAL INTERACTION AND THEIR TYPES // PLANT MIC...
MICROBIAL INTERACTION PPT/ MICROBIAL INTERACTION AND THEIR TYPES // PLANT MIC...ABHISHEK SONI NIMT INSTITUTE OF MEDICAL AND PARAMEDCIAL SCIENCES , GOVT PG COLLEGE NOIDA
Microbial interaction
Microorganisms interacts with each other and can be physically associated with another organisms in a variety of ways.
One organism can be located on the surface of another organism as an ectobiont or located within another organism as endobiont.
Microbial interaction may be positive such as mutualism, proto-cooperation, commensalism or may be negative such as parasitism, predation or competition
Types of microbial interaction
Positive interaction: mutualism, proto-cooperation, commensalism
Negative interaction: Ammensalism (antagonism), parasitism, predation, competition
I. Mutualism:
It is defined as the relationship in which each organism in interaction gets benefits from association. It is an obligatory relationship in which mutualist and host are metabolically dependent on each other.
Mutualistic relationship is very specific where one member of association cannot be replaced by another species.
Mutualism require close physical contact between interacting organisms.
Relationship of mutualism allows organisms to exist in habitat that could not occupied by either species alone.
Mutualistic relationship between organisms allows them to act as a single organism.
Examples of mutualism:
i. Lichens:
Lichens are excellent example of mutualism.
They are the association of specific fungi and certain genus of algae. In lichen, fungal partner is called mycobiont and algal partner is called
II. Syntrophism:
It is an association in which the growth of one organism either depends on or improved by the substrate provided by another organism.
In syntrophism both organism in association gets benefits.
Compound A
Utilized by population 1
Compound B
Utilized by population 2
Compound C
utilized by both Population 1+2
Products
In this theoretical example of syntrophism, population 1 is able to utilize and metabolize compound A, forming compound B but cannot metabolize beyond compound B without co-operation of population 2. Population 2is unable to utilize compound A but it can metabolize compound B forming compound C. Then both population 1 and 2 are able to carry out metabolic reaction which leads to formation of end product that neither population could produce alone.
Examples of syntrophism:
i. Methanogenic ecosystem in sludge digester
Methane produced by methanogenic bacteria depends upon interspecies hydrogen transfer by other fermentative bacteria.
Anaerobic fermentative bacteria generate CO2 and H2 utilizing carbohydrates which is then utilized by methanogenic bacteria (Methanobacter) to produce methane.
ii. Lactobacillus arobinosus and Enterococcus faecalis:
In the minimal media, Lactobacillus arobinosus and Enterococcus faecalis are able to grow together but not alone.
The synergistic relationship between E. faecalis and L. arobinosus occurs in which E. faecalis require folic acid
Describing and Interpreting an Immersive Learning Case with the Immersion Cub...
Current descriptions of immersive learning cases are often difficult or impossible to compare. This is due to a myriad of different options on what details to include, which aspects are relevant, and on the descriptive approaches employed. Also, these aspects often combine very specific details with more general guidelines or indicate intents and rationales without clarifying their implementation. In this paper we provide a method to describe immersive learning cases that is structured to enable comparisons, yet flexible enough to allow researchers and practitioners to decide which aspects to include. This method leverages a taxonomy that classifies educational aspects at three levels (uses, practices, and strategies) and then utilizes two frameworks, the Immersive Learning Brain and the Immersion Cube, to enable a structured description and interpretation of immersive learning cases. The method is then demonstrated on a published immersive learning case on training for wind turbine maintenance using virtual reality. Applying the method results in a structured artifact, the Immersive Learning Case Sheet, that tags the case with its proximal uses, practices, and strategies, and refines the free text case description to ensure that matching details are included. This contribution is thus a case description method in support of future comparative research of immersive learning cases. We then discuss how the resulting description and interpretation can be leveraged to change immersion learning cases, by enriching them (considering low-effort changes or additions) or innovating (exploring more challenging avenues of transformation). The method holds significant promise to support better-grounded research in immersive learning.
Evidence of Jet Activity from the Secondary Black Hole in the OJ 287 Binary S...
Wereport the study of a huge optical intraday flare on 2021 November 12 at 2 a.m. UT in the blazar OJ287. In the binary black hole model, it is associated with an impact of the secondary black hole on the accretion disk of the primary. Our multifrequency observing campaign was set up to search for such a signature of the impact based on a prediction made 8 yr earlier. The first I-band results of the flare have already been reported by Kishore et al. (2024). Here we combine these data with our monitoring in the R-band. There is a big change in the R–I spectral index by 1.0 ±0.1 between the normal background and the flare, suggesting a new component of radiation. The polarization variation during the rise of the flare suggests the same. The limits on the source size place it most reasonably in the jet of the secondary BH. We then ask why we have not seen this phenomenon before. We show that OJ287 was never before observed with sufficient sensitivity on the night when the flare should have happened according to the binary model. We also study the probability that this flare is just an oversized example of intraday variability using the Krakow data set of intense monitoring between 2015 and 2023. We find that the occurrence of a flare of this size and rapidity is unlikely. In machine-readable Tables 1 and 2, we give the full orbit-linked historical light curve of OJ287 as well as the dense monitoring sample of Krakow.
在线办理(salfor毕业证书)索尔福德大学毕业证毕业完成信一模一样
学校原件一模一样【微信:741003700 】《(salfor毕业证书)索尔福德大学毕业证》【微信:741003700 】学位证,留信认证(真实可查,永久存档)原件一模一样纸张工艺/offer、雅思、外壳等材料/诚信可靠,可直接看成品样本,帮您解决无法毕业带来的各种难题!外壳,原版制作,诚信可靠,可直接看成品样本。行业标杆!精益求精,诚心合作,真诚制作!多年品质 ,按需精细制作,24小时接单,全套进口原装设备。十五年致力于帮助留学生解决难题,包您满意。
本公司拥有海外各大学样板无数,能完美还原。
1:1完美还原海外各大学毕业材料上的工艺:水印,阴影底纹,钢印LOGO烫金烫银,LOGO烫金烫银复合重叠。文字图案浮雕、激光镭射、紫外荧光、温感、复印防伪等防伪工艺。材料咨询办理、认证咨询办理请加学历顾问Q/微741003700
【主营项目】
一.毕业证【q微741003700】成绩单、使馆认证、教育部认证、雅思托福成绩单、学生卡等!
二.真实使馆公证(即留学回国人员证明,不成功不收费)
三.真实教育部学历学位认证(教育部存档!教育部留服网站永久可查)
四.办理各国各大学文凭(一对一专业服务,可全程监控跟踪进度)
如果您处于以下几种情况:
◇在校期间,因各种原因未能顺利毕业……拿不到官方毕业证【q/微741003700】
◇面对父母的压力,希望尽快拿到;
◇不清楚认证流程以及材料该如何准备;
◇回国时间很长,忘记办理;
◇回国马上就要找工作,办给用人单位看;
◇企事业单位必须要求办理的
◇需要报考公务员、购买免税车、落转户口
◇申请留学生创业基金
留信网认证的作用:
1:该专业认证可证明留学生真实身份
2:同时对留学生所学专业登记给予评定
3:国家专业人才认证中心颁发入库证书
4:这个认证书并且可以归档倒地方
5:凡事获得留信网入网的信息将会逐步更新到个人身份内,将在公安局网内查询个人身份证信息后,同步读取人才网入库信息
6:个人职称评审加20分
7:个人信誉贷款加10分
8:在国家人才网主办的国家网络招聘大会中纳入资料,供国家高端企业选择人才
Farming systems analysis: what have we learnt?.pptx
Presentation given at the official farewell of Prof Ken Gillet at Wageningen on 13 June 2024
Travis Hills of MN is Making Clean Water Accessible to All Through High Flux ...
By harnessing the power of High Flux Vacuum Membrane Distillation, Travis Hills from MN envisions a future where clean and safe drinking water is accessible to all, regardless of geographical location or economic status.
The binding of cosmological structures by massless topological defects
Assuming spherical symmetry and weak field, it is shown that if one solves the Poisson equation or the Einstein field
equations sourced by a topological defect, i.e. a singularity of a very specific form, the result is a localized gravitational
field capable of driving flat rotation (i.e. Keplerian circular orbits at a constant speed for all radii) of test masses on a thin
spherical shell without any underlying mass. Moreover, a large-scale structure which exploits this solution by assembling
concentrically a number of such topological defects can establish a flat stellar or galactic rotation curve, and can also deflect
light in the same manner as an equipotential (isothermal) sphere. Thus, the need for dark matter or modified gravity theory is
mitigated, at least in part.
The cost of acquiring information by natural selection
This is a short talk that I gave at the Banff International Research Station workshop on Modeling and Theory in Population Biology. The idea is to try to understand how the burden of natural selection relates to the amount of information that selection puts into the genome.
It's based on the first part of this research paper:
The cost of information acquisition by natural selection
Ryan Seamus McGee, Olivia Kosterlitz, Artem Kaznatcheev, Benjamin Kerr, Carl T. Bergstrom
bioRxiv 2022.07.02.498577; doi: https://doi.org/10.1101/2022.07.02.498577
Direct Seeded Rice - Climate Smart Agriculture
Direct Seeded Rice - Climate Smart AgricultureInternational Food Policy Research Institute- South Asia Office
PPT on Direct Seeded Rice presented at the three-day 'Training and Validation Workshop on Modules of Climate Smart Agriculture (CSA) Technologies in South Asia' workshop on April 22, 2024.
Applied Science: Thermodynamics, Laws & Methodology.pdf
When I was asked to give a companion lecture in support of ‘The Philosophy of Science’ (https://shorturl.at/4pUXz) I decided not to walk through the detail of the many methodologies in order of use. Instead, I chose to employ a long standing, and ongoing, scientific development as an exemplar. And so, I chose the ever evolving story of Thermodynamics as a scientific investigation at its best.
Conducted over a period of >200 years, Thermodynamics R&D, and application, benefitted from the highest levels of professionalism, collaboration, and technical thoroughness. New layers of application, methodology, and practice were made possible by the progressive advance of technology. In turn, this has seen measurement and modelling accuracy continually improved at a micro and macro level.
Perhaps most importantly, Thermodynamics rapidly became a primary tool in the advance of applied science/engineering/technology, spanning micro-tech, to aerospace and cosmology. I can think of no better a story to illustrate the breadth of scientific methodologies and applications at their best.
Gadgets for management of stored product pests_Dr.UPR.pdf
Insectsplayamajorroleinthedeteriorationoffoodgrainscausingbothquantitativeandqualitativelosses
Wellprovedthatnogranariescanbefilledwithgrainswithoutinsectsastheharvestedproducecontainegg(or)larvae(or)pupae(or)adultinsectinthembecauseoffieldcarryoverinfestationwhichcannotbeavoidedindevelopingcountrieslikeIndia
Simpletechnologiesfortimelydetectionofinsectsinthestoredproduceandtherebyplantimelycontrolmeasures
The debris of the ‘last major merger’ is dynamically young
The Milky Way’s (MW) inner stellar halo contains an [Fe/H]-rich component with highly eccentric orbits, often referred to as the
‘last major merger.’ Hypotheses for the origin of this component include Gaia-Sausage/Enceladus (GSE), where the progenitor
collided with the MW proto-disc 8–11 Gyr ago, and the Virgo Radial Merger (VRM), where the progenitor collided with the
MW disc within the last 3 Gyr. These two scenarios make different predictions about observable structure in local phase space,
because the morphology of debris depends on how long it has had to phase mix. The recently identified phase-space folds in Gaia
DR3 have positive caustic velocities, making them fundamentally different than the phase-mixed chevrons found in simulations
at late times. Roughly 20 per cent of the stars in the prograde local stellar halo are associated with the observed caustics. Based
on a simple phase-mixing model, the observed number of caustics are consistent with a merger that occurred 1–2 Gyr ago.
We also compare the observed phase-space distribution to FIRE-2 Latte simulations of GSE-like mergers, using a quantitative
measurement of phase mixing (2D causticality). The observed local phase-space distribution best matches the simulated data
1–2 Gyr after collision, and certainly not later than 3 Gyr. This is further evidence that the progenitor of the ‘last major merger’
did not collide with the MW proto-disc at early times, as is thought for the GSE, but instead collided with the MW disc within
the last few Gyr, consistent with the body of work surrounding the VRM.
Recently uploaded (20)
Mending Clothing to Support Sustainable Fashion_CIMaR 2024.pdf
Mending Clothing to Support Sustainable Fashion_CIMaR 2024.pdf
MICROBIAL INTERACTION PPT/ MICROBIAL INTERACTION AND THEIR TYPES // PLANT MIC...
MICROBIAL INTERACTION PPT/ MICROBIAL INTERACTION AND THEIR TYPES // PLANT MIC...
Describing and Interpreting an Immersive Learning Case with the Immersion Cub...
Describing and Interpreting an Immersive Learning Case with the Immersion Cub...
Evidence of Jet Activity from the Secondary Black Hole in the OJ 287 Binary S...
Evidence of Jet Activity from the Secondary Black Hole in the OJ 287 Binary S...
Farming systems analysis: what have we learnt?.pptx
Farming systems analysis: what have we learnt?.pptx
Travis Hills of MN is Making Clean Water Accessible to All Through High Flux ...
Travis Hills of MN is Making Clean Water Accessible to All Through High Flux ...
The binding of cosmological structures by massless topological defects
The binding of cosmological structures by massless topological defects
The cost of acquiring information by natural selection
The cost of acquiring information by natural selection
Applied Science: Thermodynamics, Laws & Methodology.pdf
Applied Science: Thermodynamics, Laws & Methodology.pdf
Microbiology of Central Nervous System INFECTIONS.pdf
Microbiology of Central Nervous System INFECTIONS.pdf
Gadgets for management of stored product pests_Dr.UPR.pdf
Gadgets for management of stored product pests_Dr.UPR.pdf
The debris of the ‘last major merger’ is dynamically young
The debris of the ‘last major merger’ is dynamically young
Protein structure prediction (1)
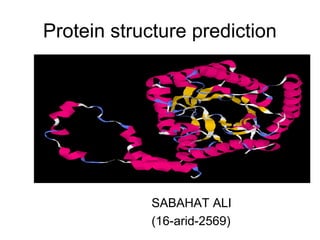
- 1. Protein structure prediction SABAHAT ALI (16-arid-2569)
- 2. Introduction • Proteins are one of the most important parts in any biological systems. • Understanding the folding of the amino- acid chain to produce functional proteins is essential for studying cellular systems. • Fast and accessible methods of solving the 3D structure of a protein are in high demand.
- 3. Protein structure • This topic has been covered several times. Next!
- 4. Computational methods • Ab initio- methods – Laws of physics + amino-acid sequence = protein structure – Computes potential energy functions. – Minimum potential energy is the most stable structure and as such the most likely. – Computationally demanding.
- 5. Comparative methods • Based on the limited amount of possible tertiary structure types. • Approximately 2000 different types of protein folds. • Comparing the sample to a database of known structures, for example Protein Data Bank.
- 6. Homology modelling • Based on the assumption that homologous (related) proteins fold in a similar fashion. • Folding is a highly conserved factor, much more so than amino-acid sequence. • Finding a match between two distantly related proteins can be difficult.
- 7. Protein threading prtactical • Based on the assumption that similar folding has already been found. • Comparing parts of the sequence to a database of known three dimensional structures using a scoring function. • Works at least somewhat on approximately 80% of new protein sequences.
- 8. The End