Downloaded 115 times

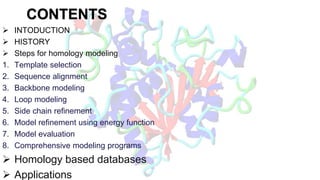


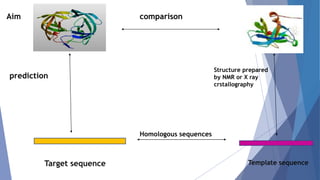

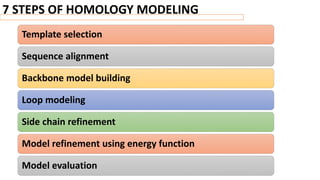
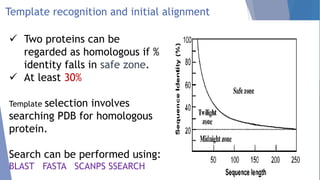


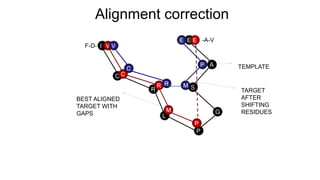
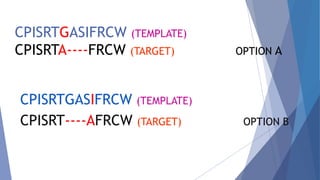



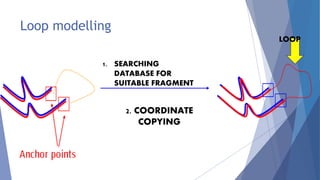

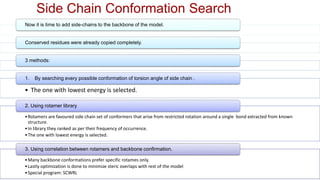


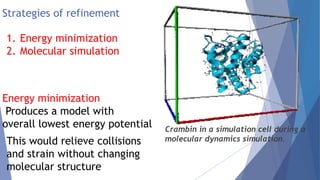










Homology modeling is a computational method to predict the 3D structure of a protein based on the known structure of homologous proteins. It involves 7 main steps: 1) selecting a template protein with high sequence similarity, 2) aligning the sequences, 3) building the protein backbone, 4) modeling loops and insertions/deletions, 5) refining side chains, 6) refining the overall structure using energy minimization, and 7) evaluating the model. Homology models can accurately predict protein structure when the sequence identity between the target and template is above 30%. Models are useful for studying protein function and designing drugs.























































































