Downloaded 137 times











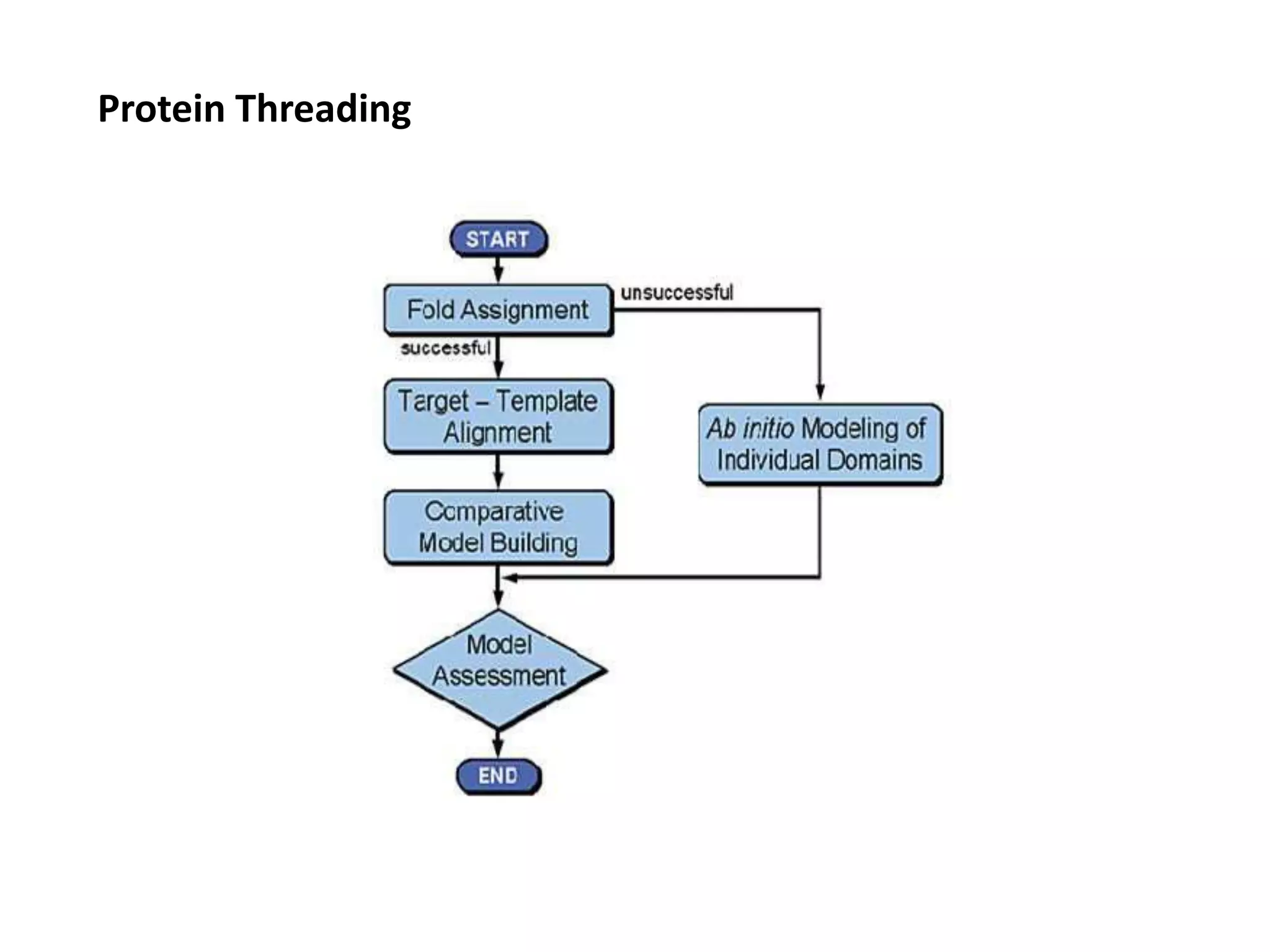
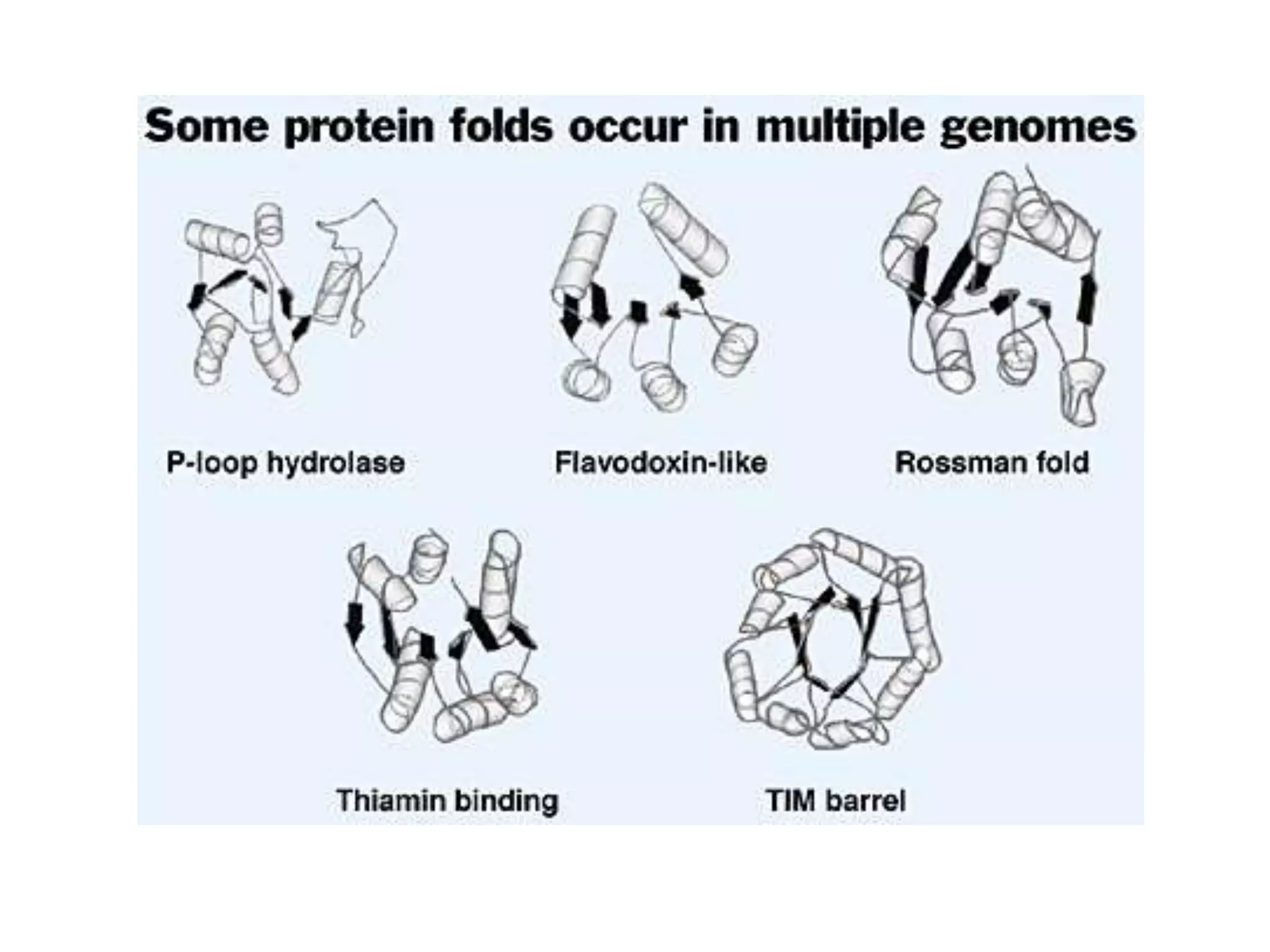
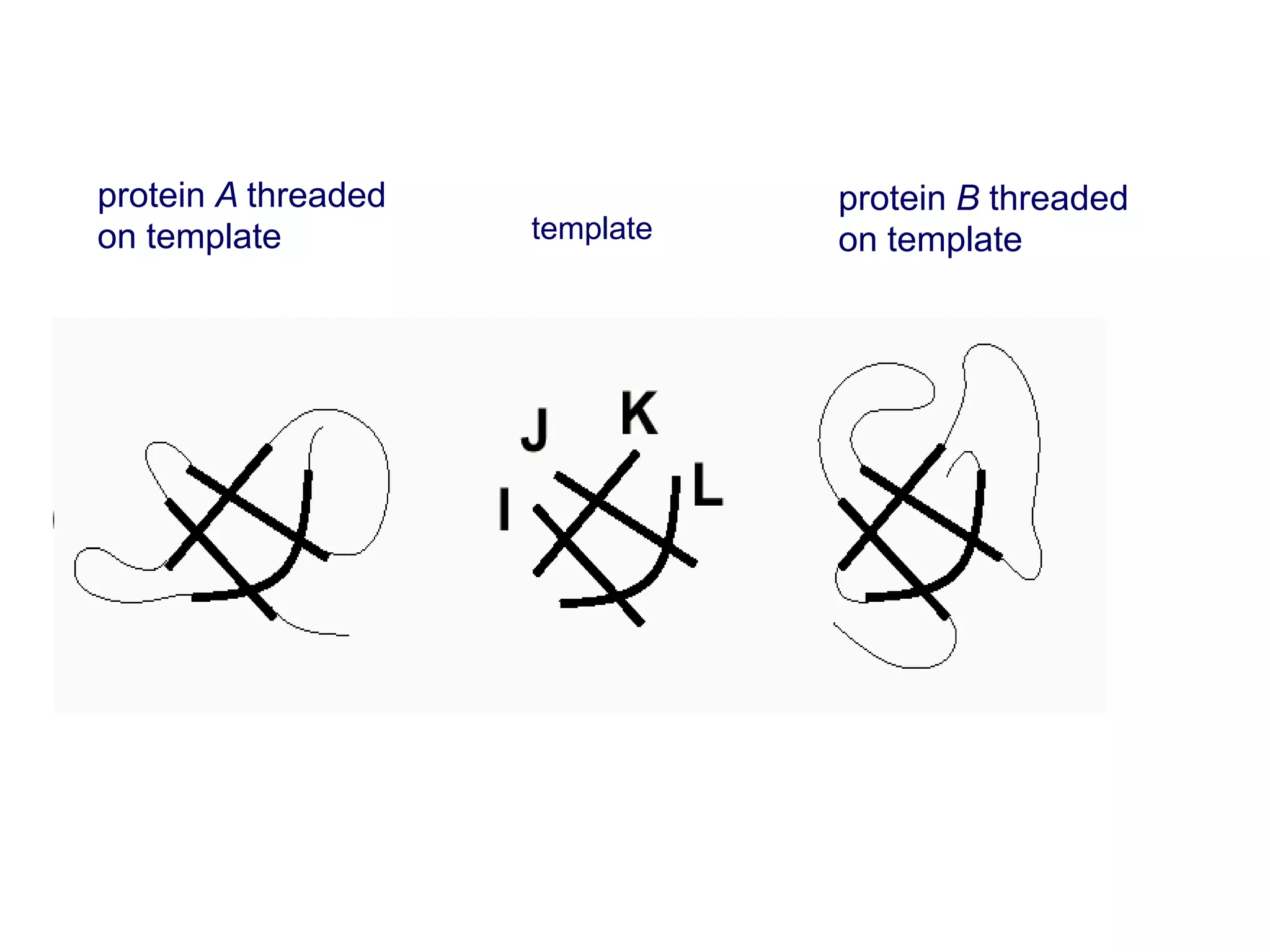

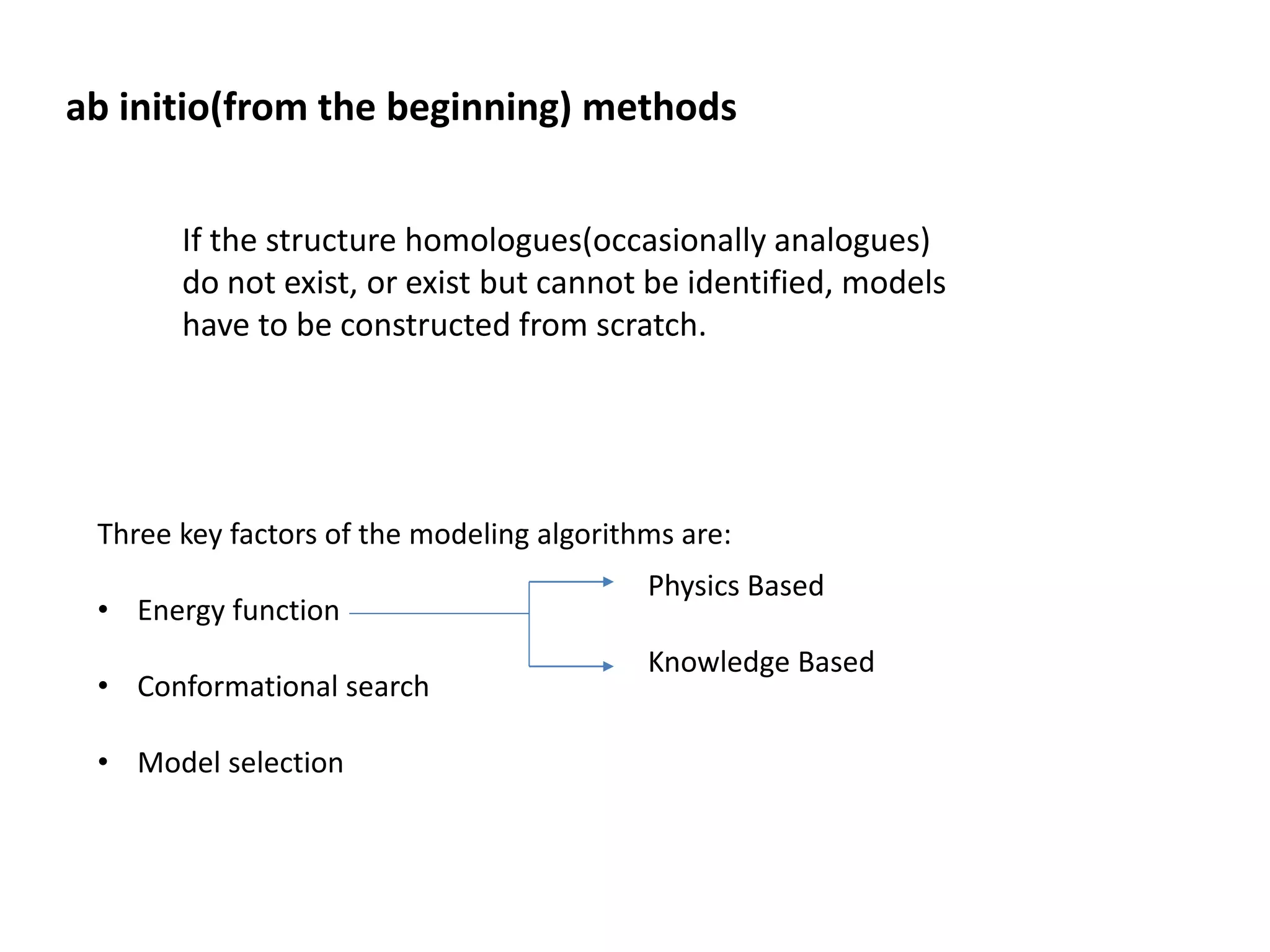






This document presents an overview of molecular modeling techniques. It discusses the history of molecular modeling and some common computational methods like molecular mechanics, quantum mechanics and molecular dynamics. It also describes different modeling approaches like template modeling techniques such as homology modeling and threading as well as template-free modeling methods including ab initio and knowledge-based modeling. The document concludes that molecular modeling can provide useful insights for research if used carefully while also noting current limitations, especially for modeling larger protein structures.











































































