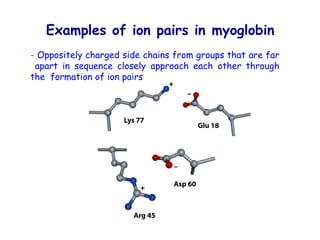
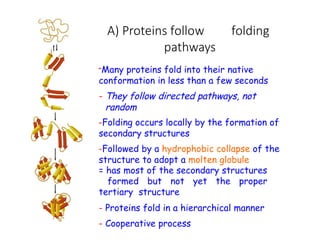
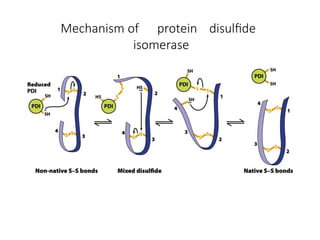
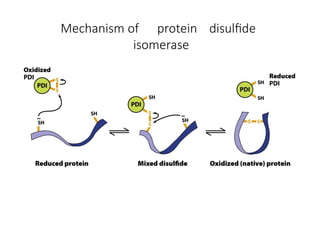
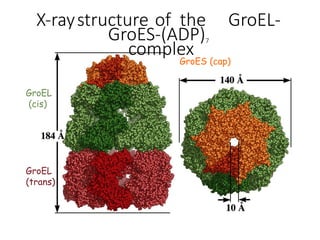
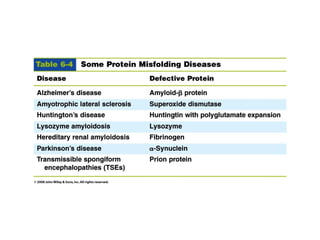
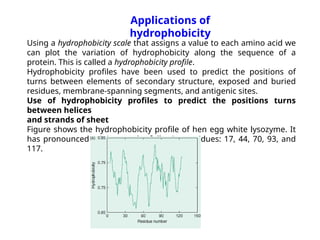
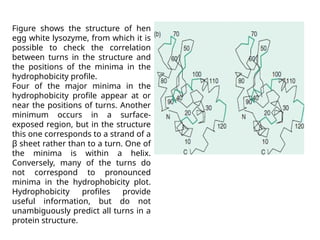
The document discusses protein stability and folding, highlighting the delicate balance of forces that maintain native protein structures, including hydrophobic effects, electrostatic interactions, and the role of disulfide bonds. It further explains the mechanisms of protein folding, emphasizing the importance of molecular chaperones and the hierarchical nature of the folding pathway, which prevents misfolding and aggregation. Additionally, applications of hydrophobicity profiles in predicting protein structure and the significance of structural similarity in protein analysis are also addressed.
















![Structural alignment of bovine γ-chymotrypsin and S.
aureus epidermolytic toxin A
Bovine chymotrypsin and S. aureus epidermolytic toxin A are both
members of the chymotrypsin family of proteinases. Figure shows a
structural superposition of PDB entries 8GCH (γ-chymotrypsin) (black)
and 1AGJ (S. aureus epidermolytic toxin A) (green). The molecules share
the common chymotrypsin-family serine
proteinase folding pattern, and the Ser-His-Asp catalytic triad (ball-and-
stick).
Structural superposition of γ-chymotrypsin [8GCH] (black) and S.
aureus epidermolytic toxin A [1AGJ] (green). The sidechains of the
catalytic triads are shown. Observe that the region around the
active site is the best-conserved part of the protein.](https://image.slidesharecdn.com/proteinstabilityandfolding-240812062524-41c9d8c3/85/Protein-Stability-and-folding-pptx-pdf-25-320.jpg)








































































![谷歌留痕技术 [ 𝙩𝙤𝙥 𝟮𝟯𝟯. 𝙘 𝙤𝙢 ]](https://cdn.slidesharecdn.com/ss_thumbnails/top233-260130174328-3833018c-thumbnail.jpg?width=640&height=640&fit=bounds)














