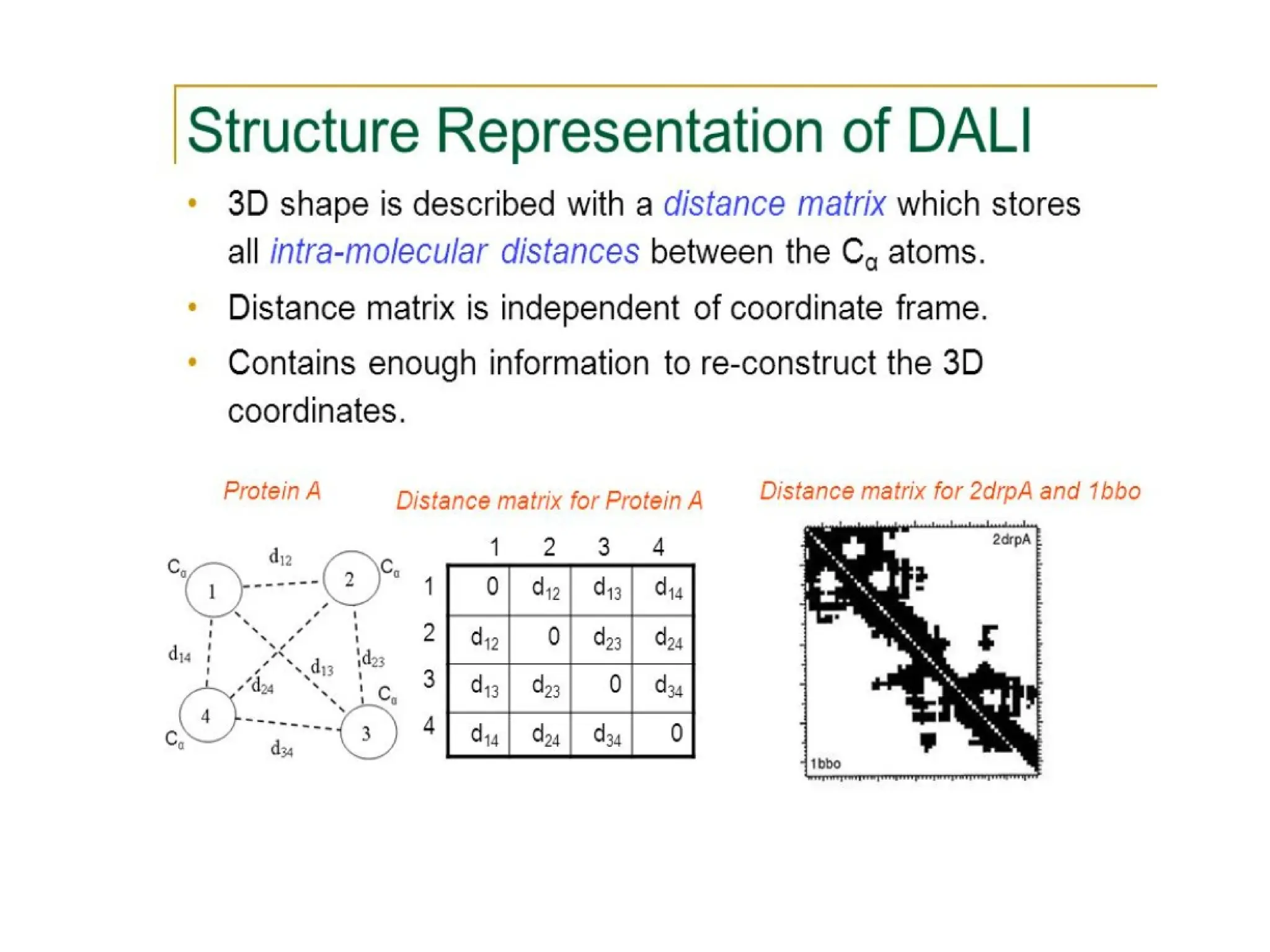
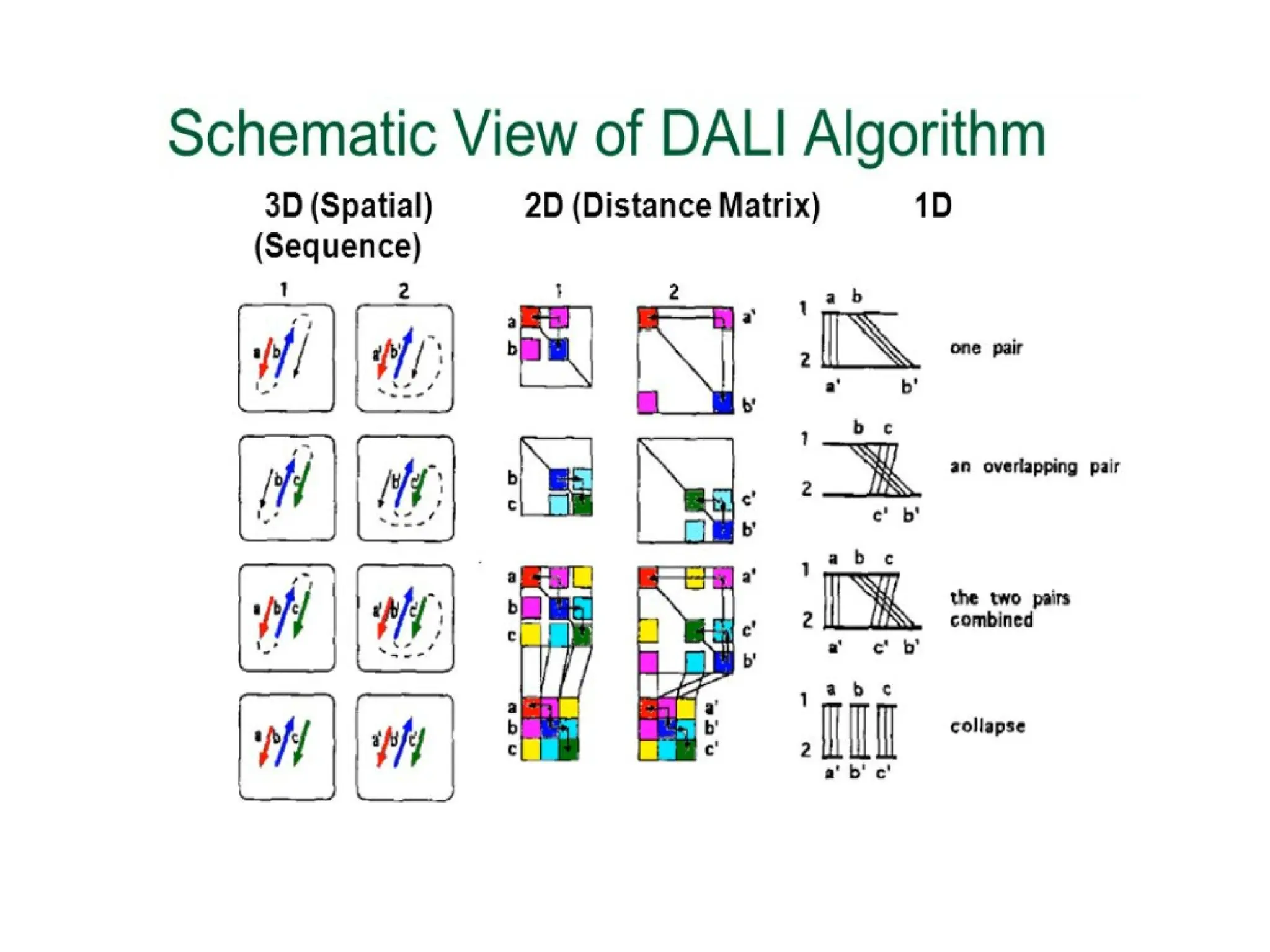
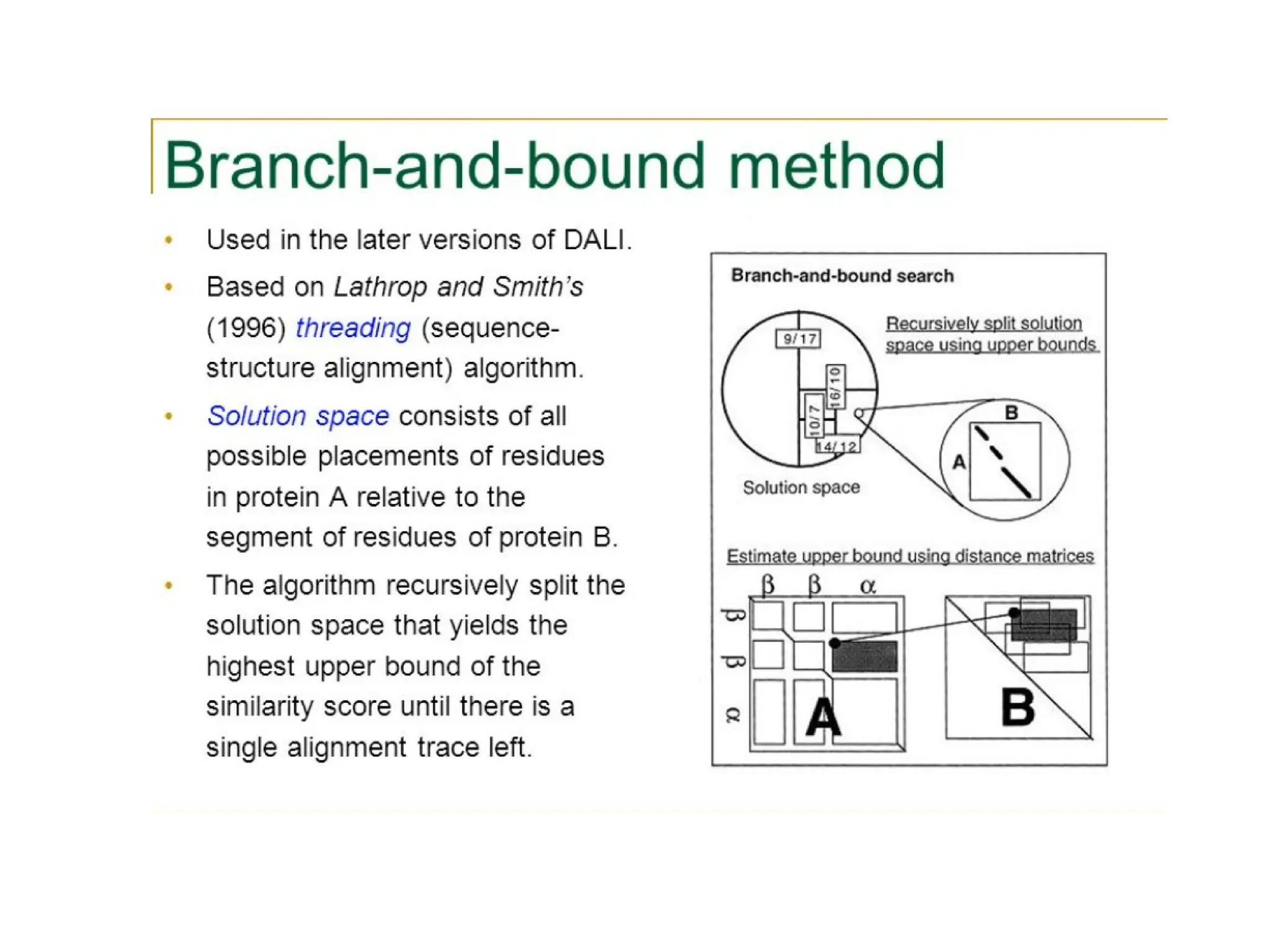
The DALI method, developed by L. Holm and C. Sander, identifies distantly related proteins by detecting conserved patterns of inter-residue contacts in protein structures. This efficient program allows for routine screening of the entire protein data bank and has uncovered significant structural similarities that are not apparent through sequence alignment. An example highlighted is the identification of structural similarities among unrelated proteins such as mouse adenosine deaminase and pseudomonas diminuta phosphotriesterase, despite having only 13% sequence identity.



![The regions of common fold, as determined by the program DALI
by L. Holm and C. Sander, in the TIM barrel proteins mouse
adenosine deaminase [1FKX] (black) and Pseudomonas diminuta
phosphotriesterase [1PTA]
(green). In the alignment shown in this figure the sequences
have only 13% identical residues](https://image.slidesharecdn.com/dalimethod-240812062522-ba0ffea9/75/Distance-matrix-ALIgnment-METHOD-pptx-pdf-4-2048.jpg)