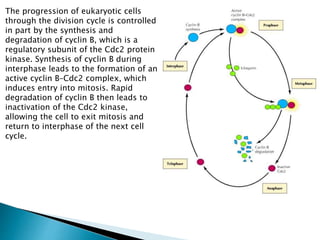
Protein folding is the process by which a polypeptide chain folds into its characteristic and functional three-dimensional structure. It involves four stages: primary structure is the amino acid sequence; secondary structure involves folding into alpha helices and beta sheets through hydrogen bonding. Tertiary structure involves folding of secondary structures into the final three-dimensional structure. Some proteins undergo quaternary structure formation through interactions between folded polypeptide chains. The folding process is driven by the hydrophobic effect and other non-covalent interactions and allows proteins to attain biologically functional conformations. Chaperone proteins assist in protein folding to facilitate efficient folding in living cells.


















![ There are two main protein quality control systems in the cell that are responsible
for eliminating protein aggregates. Misfolded proteins can get refolded by the bi-
chaperone system or degraded by the ubiquitin proteasome system or autophagy.
Refolding
The bi-chaperone system utilizes the Hsp70 (DnaK-DnaJ-GrpE in E. coli and Ssa1-
Ydj1/Sis1-Sse1/Fe1 in yeast) and Hsp100 (ClpB in E. coli and Hsp104 in yeast)
chaperones for protein disaggregation and refolding.
Hsp70 interacts with the protein aggregates and recruits Hsp100. Hsp70 stabilizes
an activated Hsp100. Hsp100 proteins have aromatic pore loops that are used for
threading activity to disentangle single polypeptides. This threading activity can
be initiated at the N-terminus, C-terminus or in the middle of the polypeptide.
The polypeptide gets translocated through Hsp100 in a series of steps, utilizing
an ATP at each step. The polypeptide unfolds and is then allowed to refold either
by itself or with the help of heat shock proteins.
Degradation[edit]
Misfolded proteins can be eliminated through the ubiquitin-proteasome system
(UPS). This consists of an E1-E2-E3 pathway that ubiquinates proteins to mark
them for degradation. In eukaryotes, the proteins get degraded by the 26S
proteasome. In mammalian cells, the E3 ligase, carboxy-terminal Hsp70
interacting protein (CHIP), targets Hsp70-bound proteins. In yeast, the E3 ligases
Doa10 and Hrd1 have similar functions on endoplasmic reticulum proteins.](https://image.slidesharecdn.com/proteinfolding-170226165229-240311170214-39d80574/85/proteinfolding-170226165229-pptx12345747-18-320.jpg)











































































![CTEV [ clubfoot] DR ARUN LAL ,DR MOHAMED ASHRAF travancore medical college k...](https://cdn.slidesharecdn.com/ss_thumbnails/ctevclubfootdrarunlaldrmohamedashraftravancoremedicalcollegekollamkeralaindia-260208063247-18fc466c-thumbnail.jpg?width=640&height=640&fit=bounds)