Downloaded 415 times
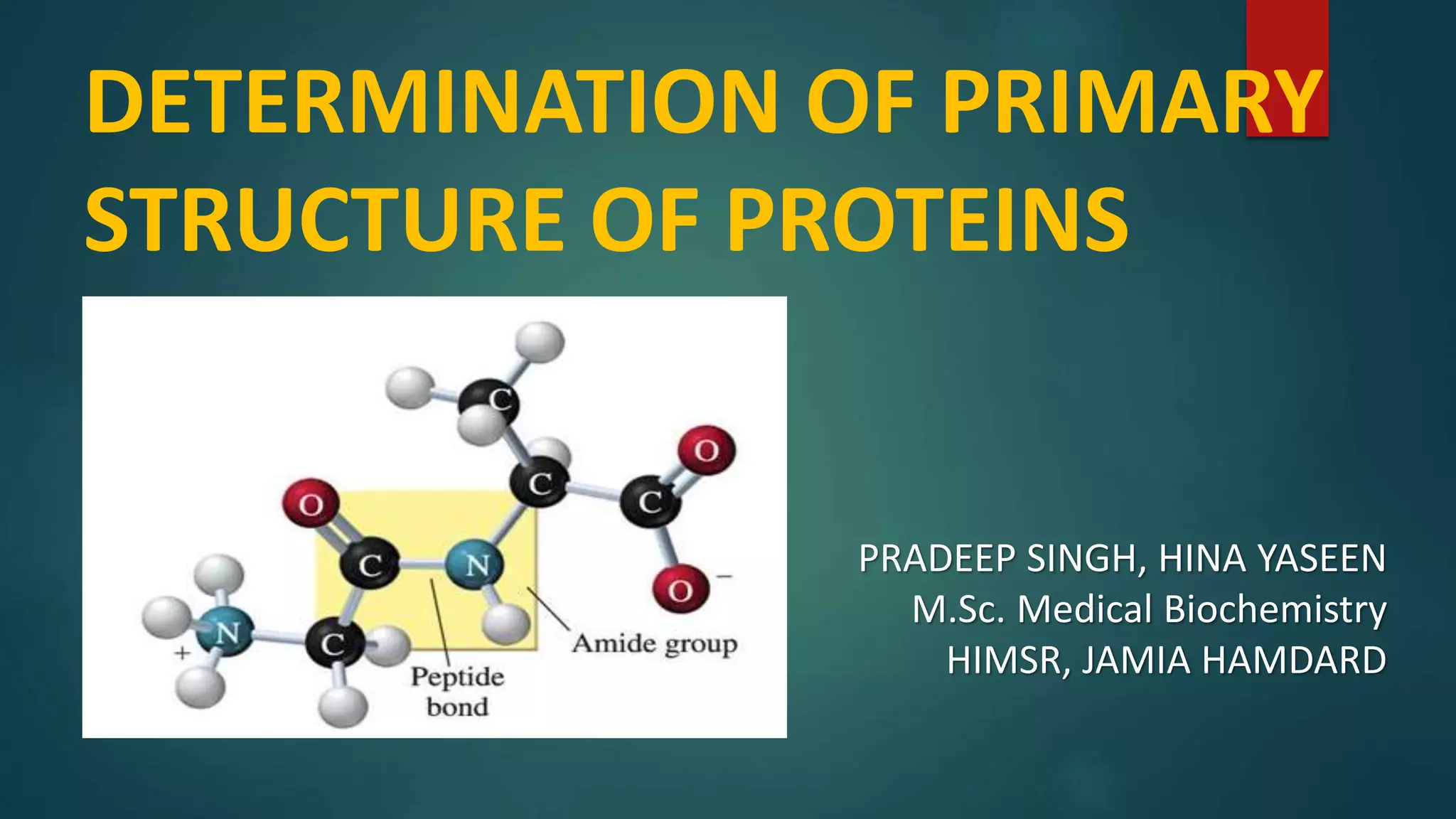




































The document discusses the determination of the primary structure of proteins. It begins by explaining that proteins are composed of amino acid residues linked by peptide bonds to form a polypeptide chain. The primary structure refers to the specific sequence of amino acids in this chain. Mass spectrometry and tandem mass spectrometry techniques are used to analyze protein fragments obtained through enzymatic or chemical cleavage to determine the amino acid sequence and thereby elucidate the primary structure.
Overview of proteins as polymers of amino acids, their significance, components, and molecular weight.
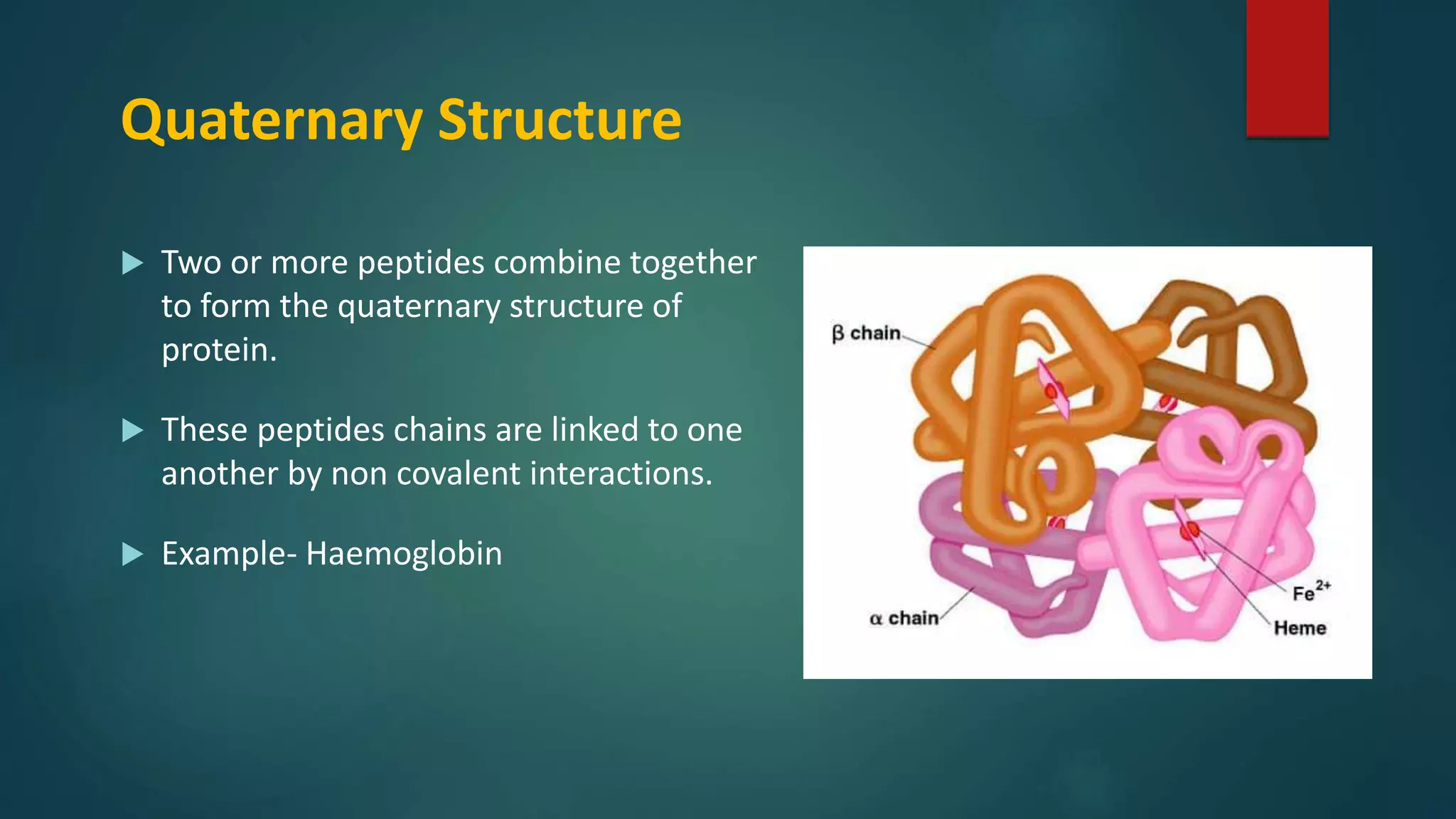
Introduction to four protein structural levels: primary, secondary, tertiary, and quaternary.
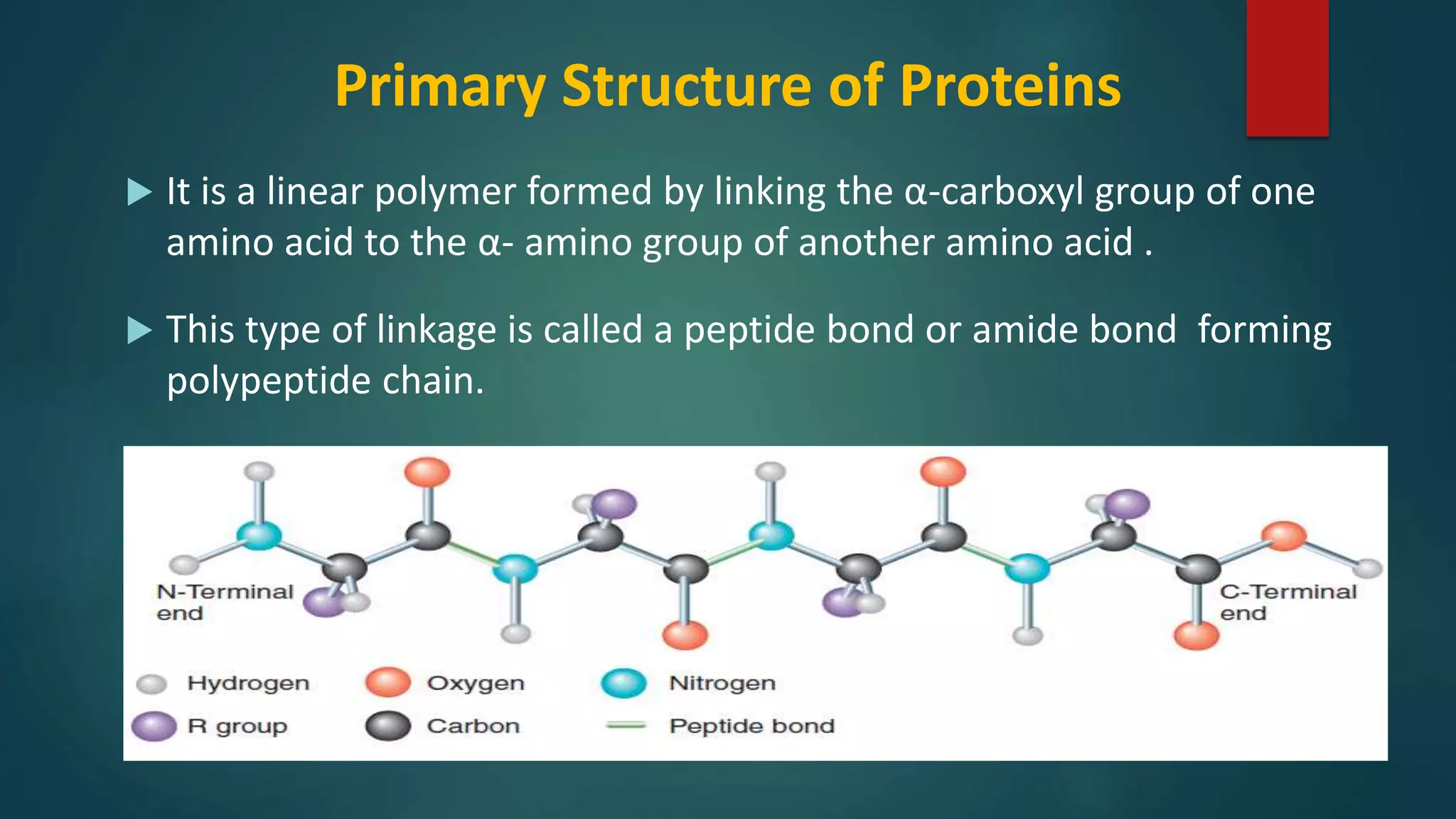
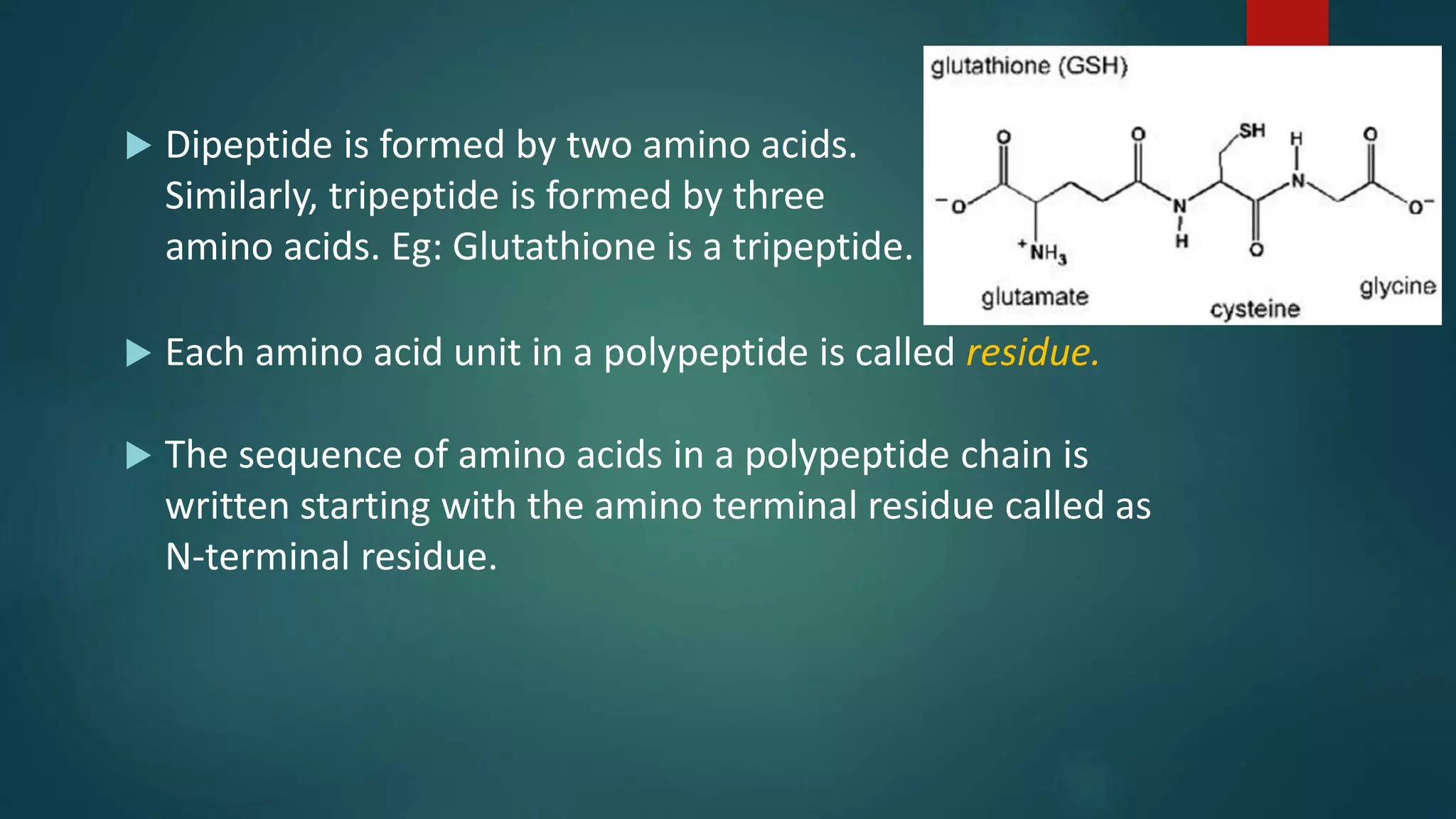
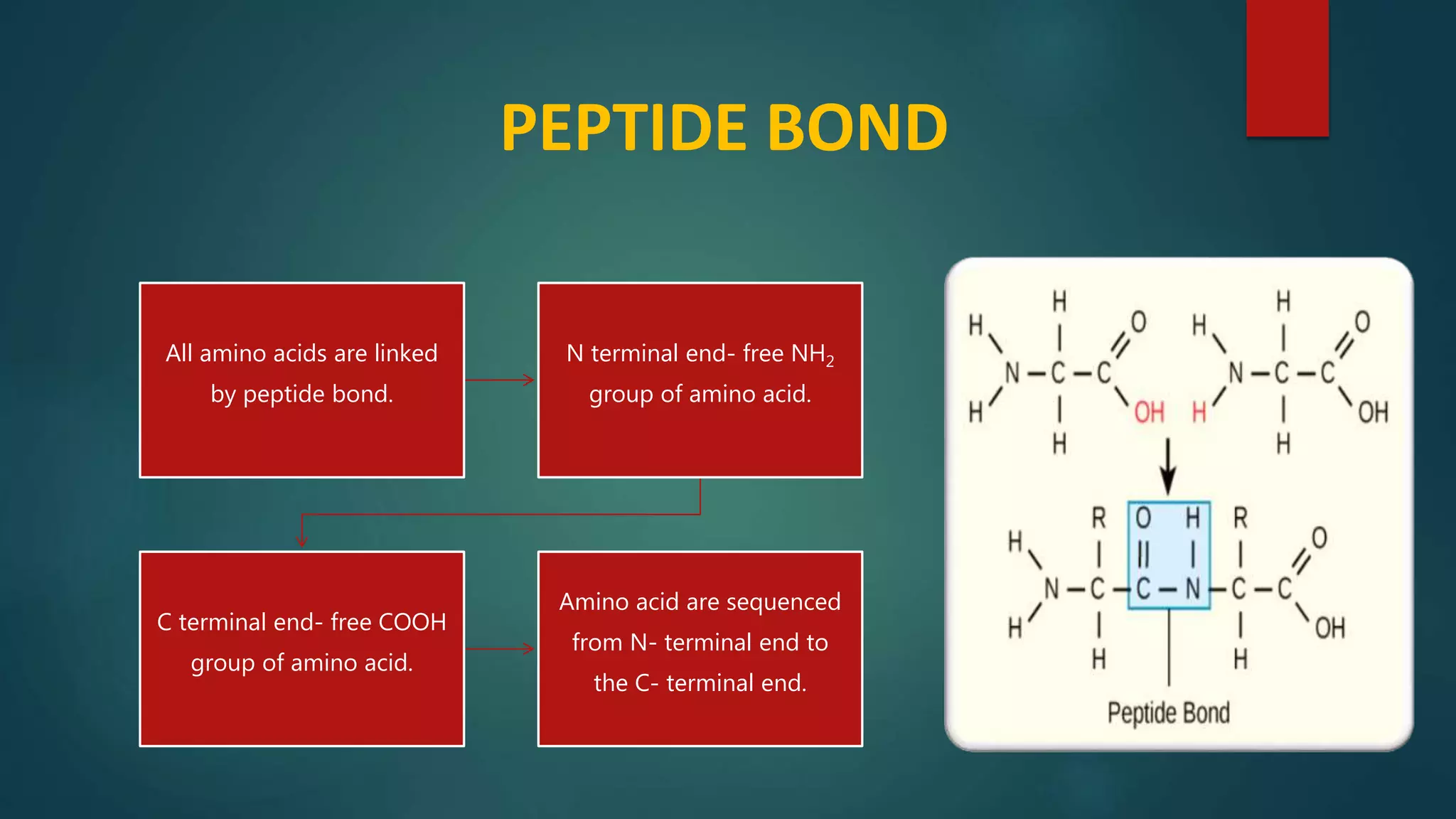
Details on primary structure, formation of polypeptides, and description of peptide bonds.
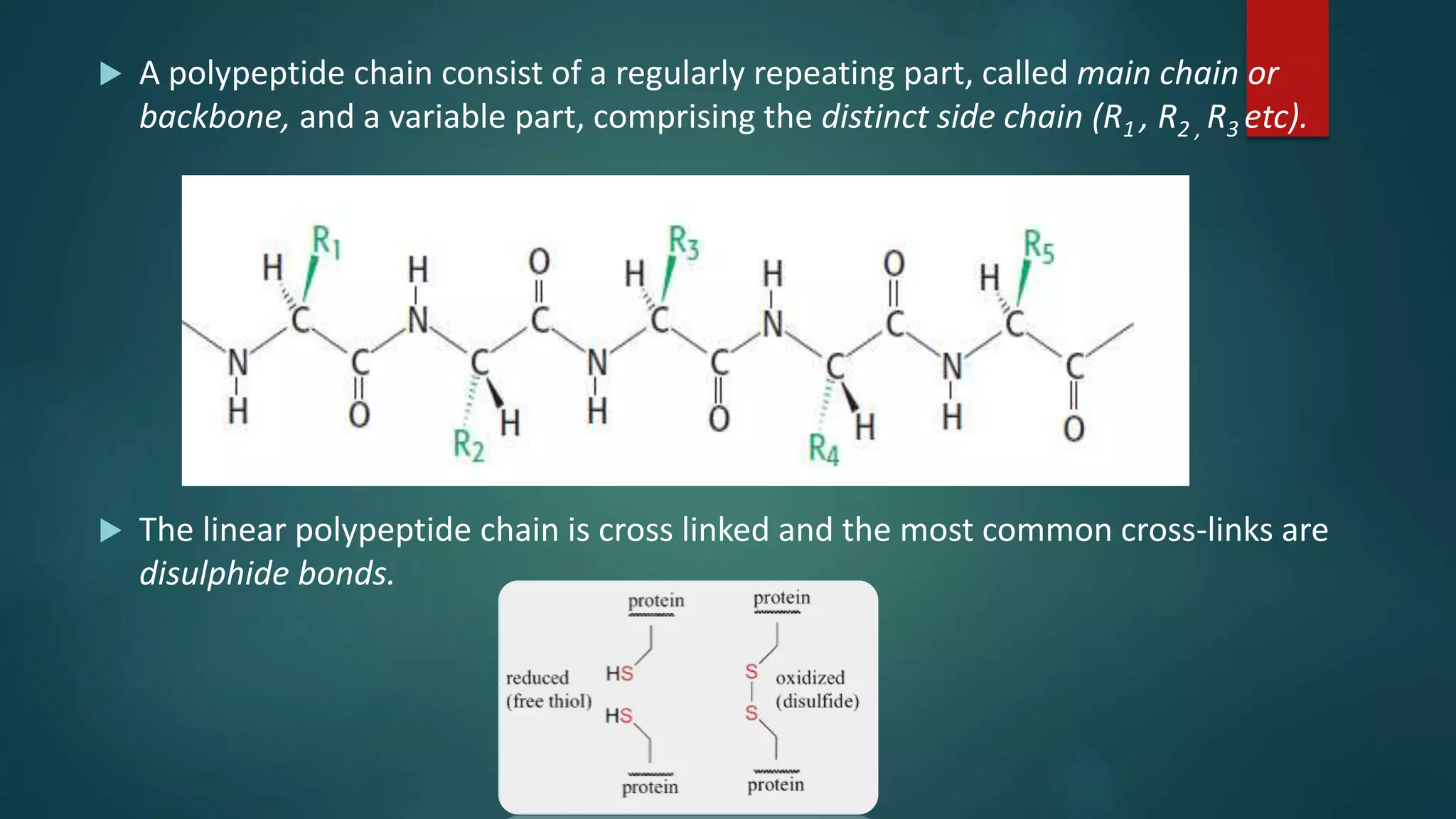
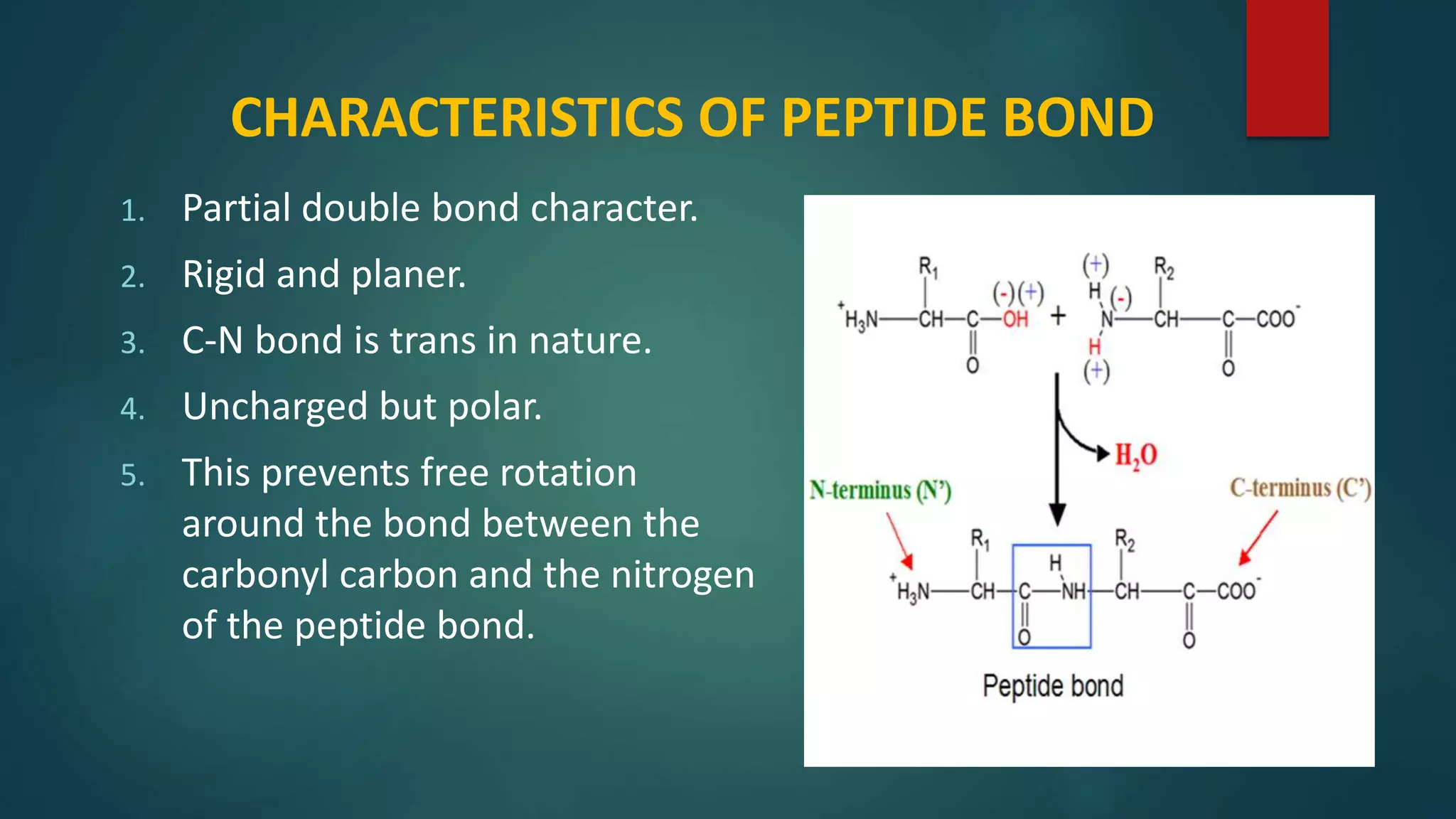
Key characteristics of peptide bonds and their structural implications in protein formation.
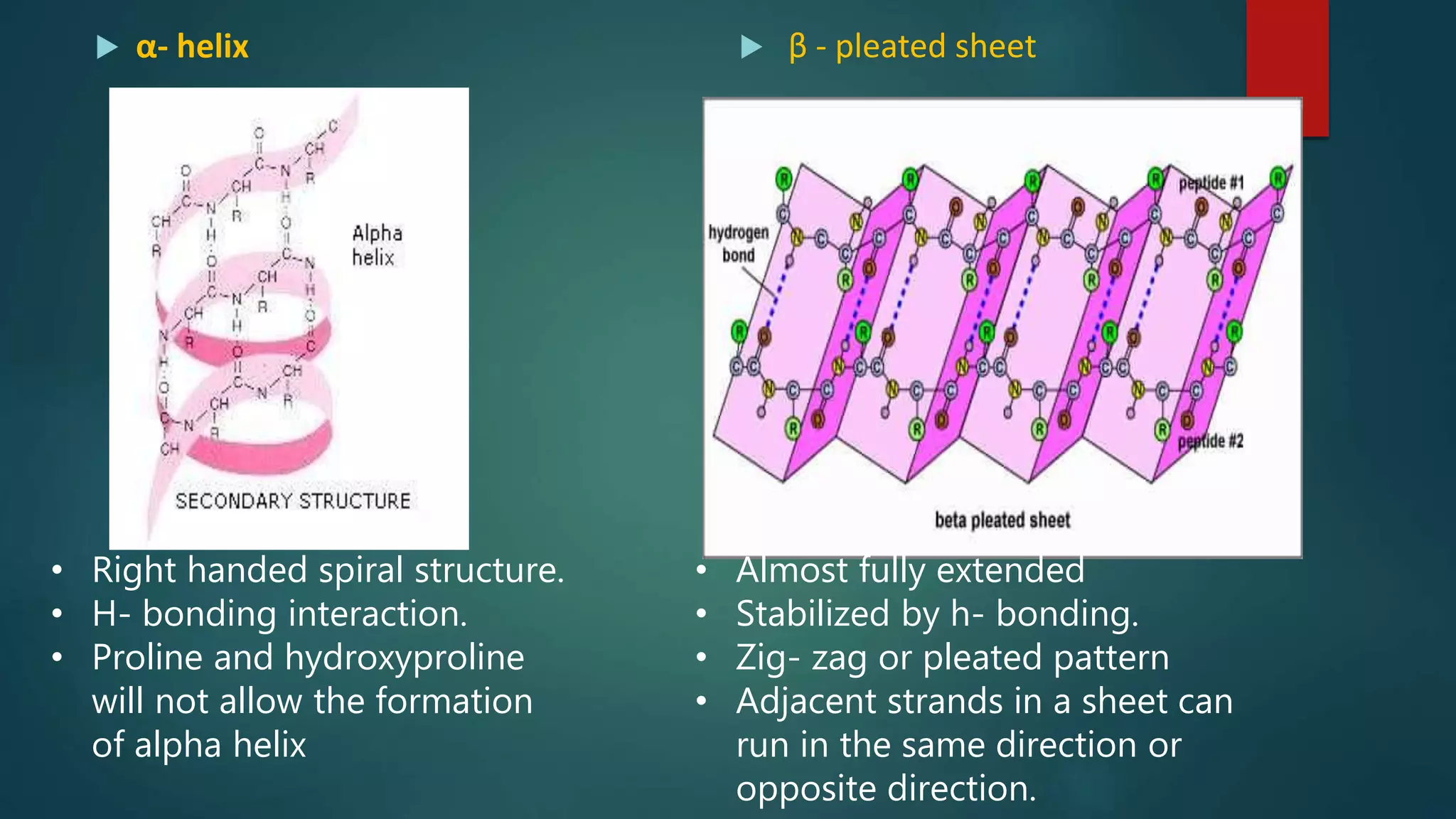
Characteristics and types of secondary structure, including alpha helices and beta sheets.
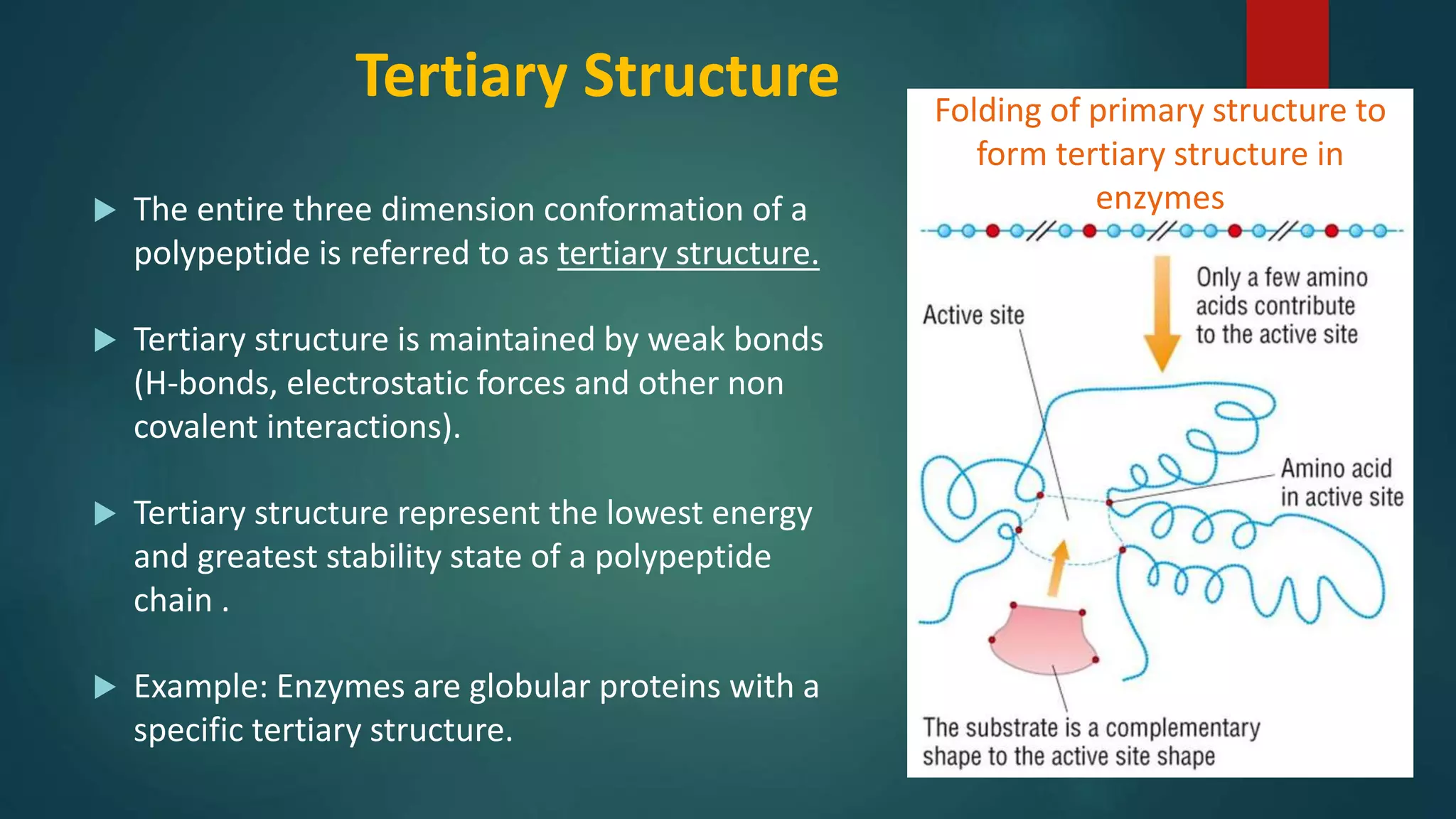
Description of tertiary structure and its stabilization mechanisms with examples of globular proteins.
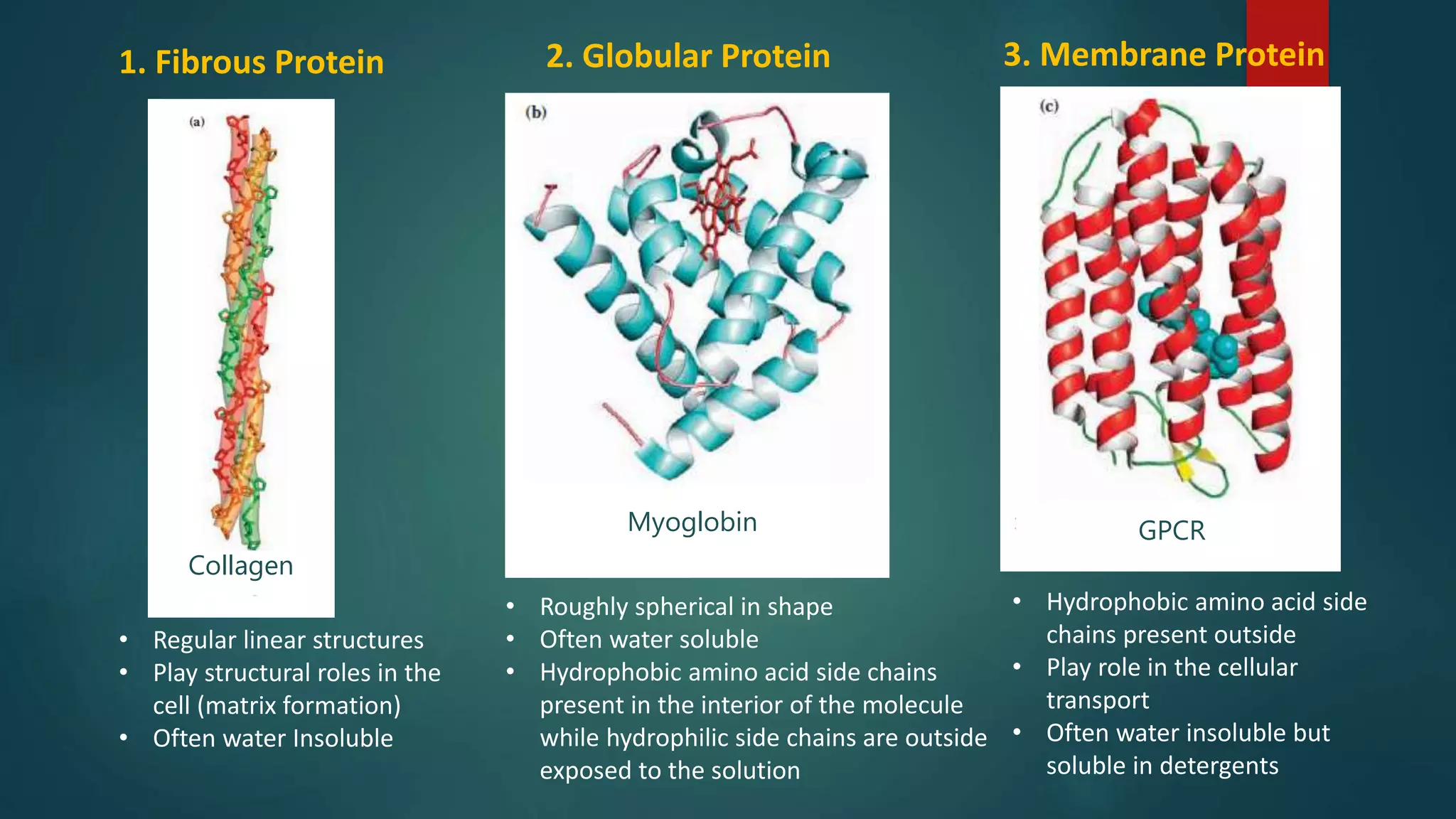
Classification of proteins based on structure: fibrous, globular, and membrane proteins.
Reasons for determining the primary structure of proteins and their biological roles.
Overview of protein purification steps, including solubilization, stabilization, and peptide digestion.
Methods to determine amino acid composition through hydrolysis and various techniques.
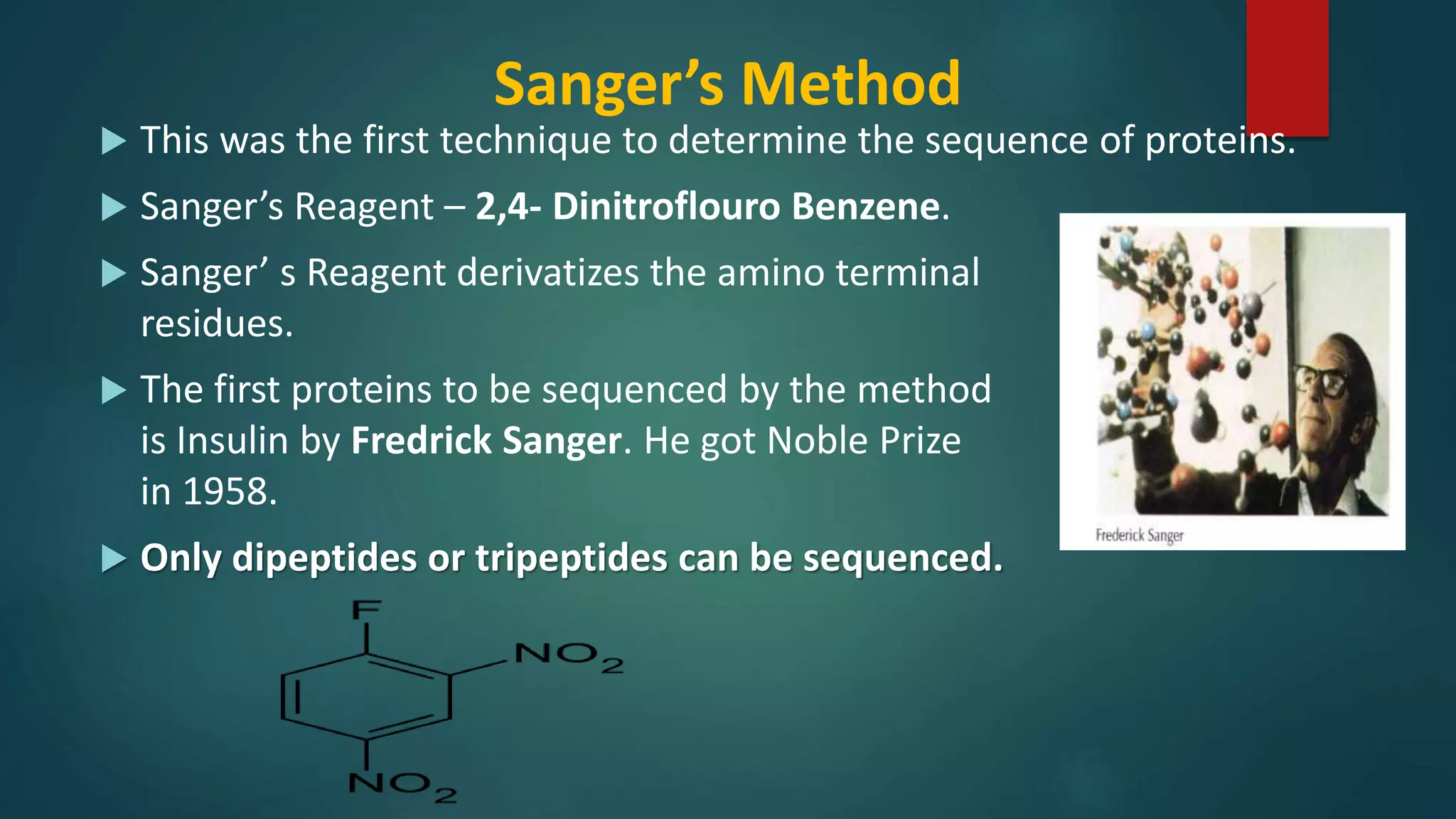
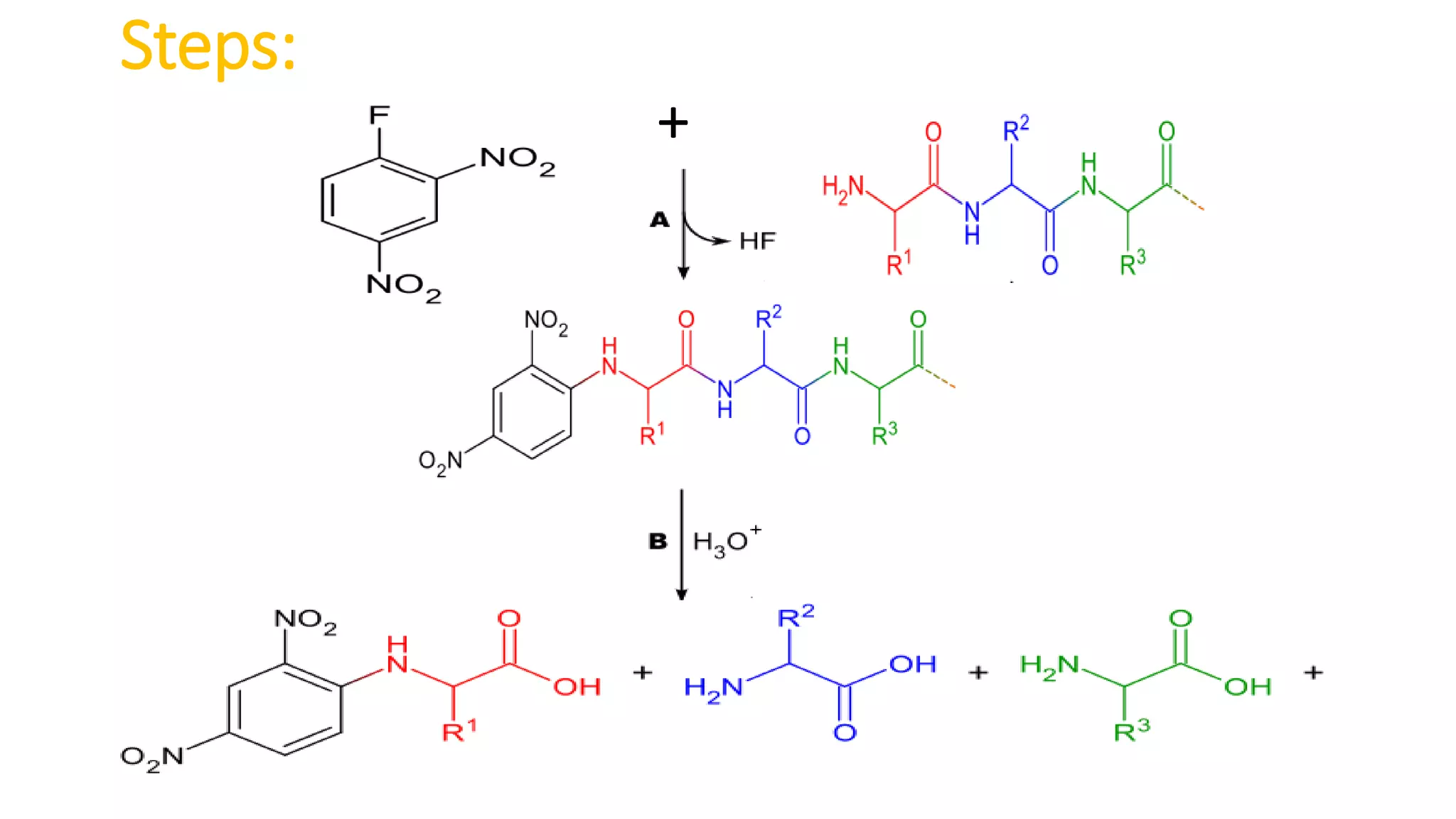
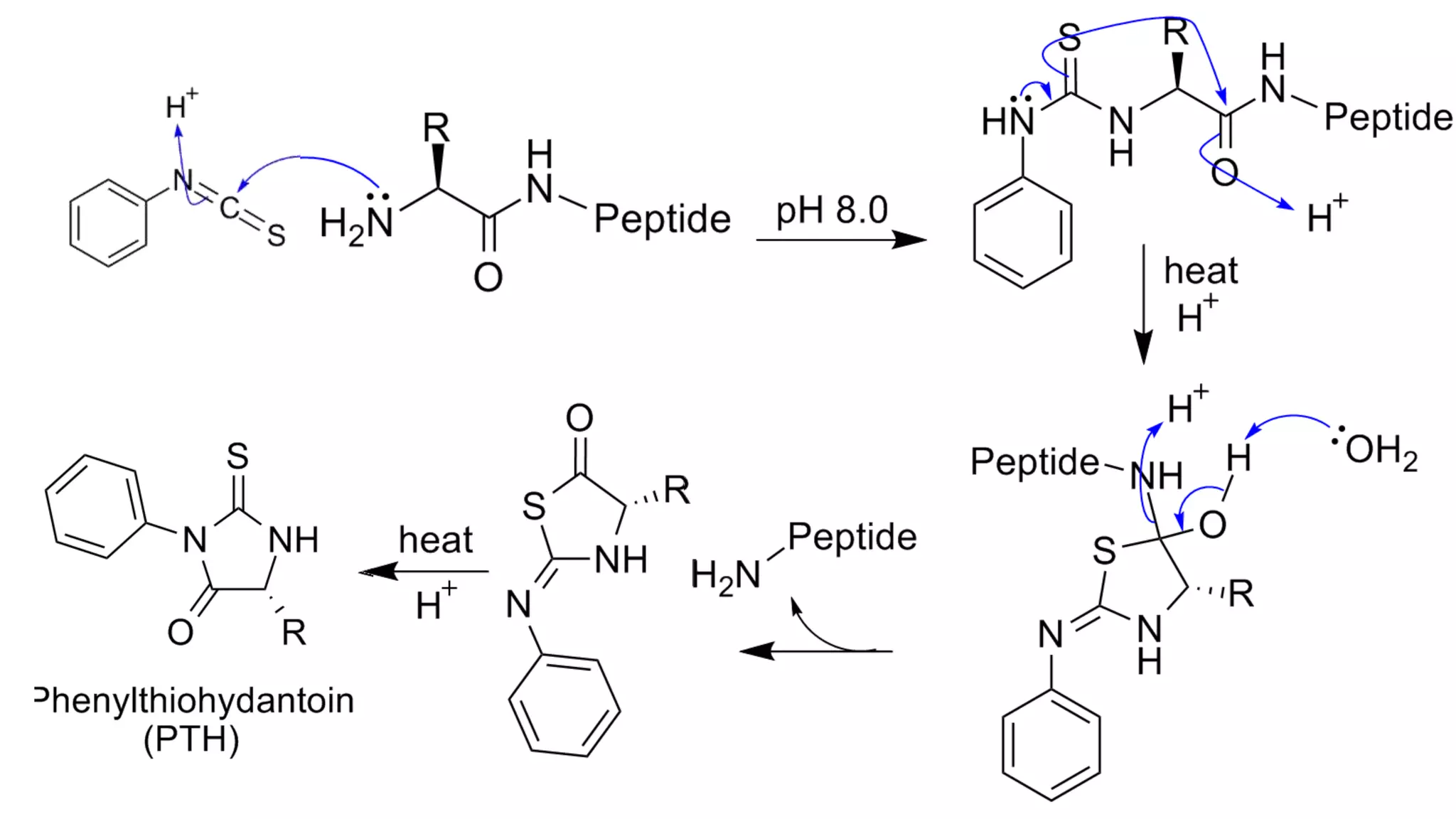
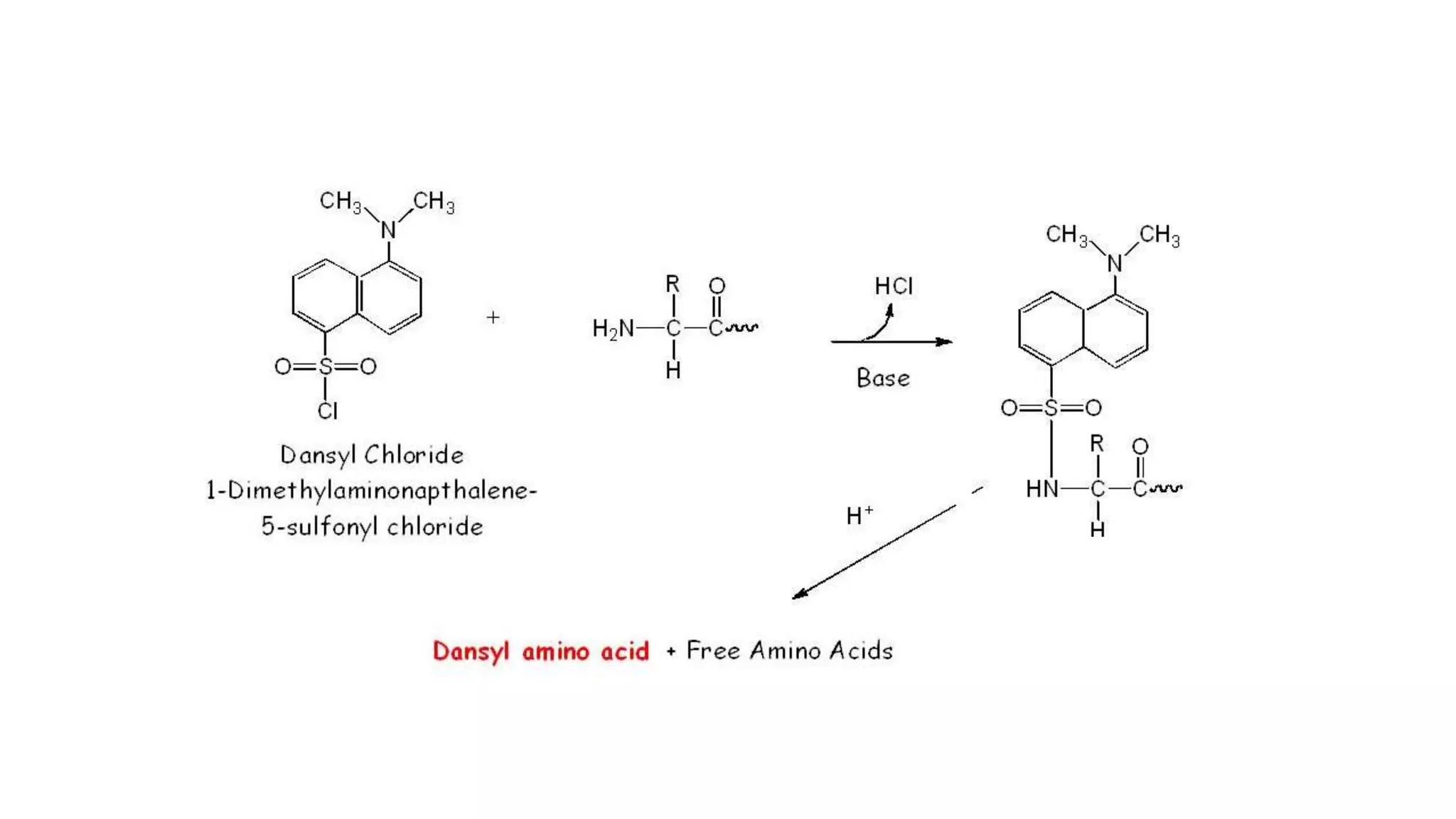
Methods for identifying N-terminal amino acids using Sanger's method, Edman's degradation, and Dansyl chloride.
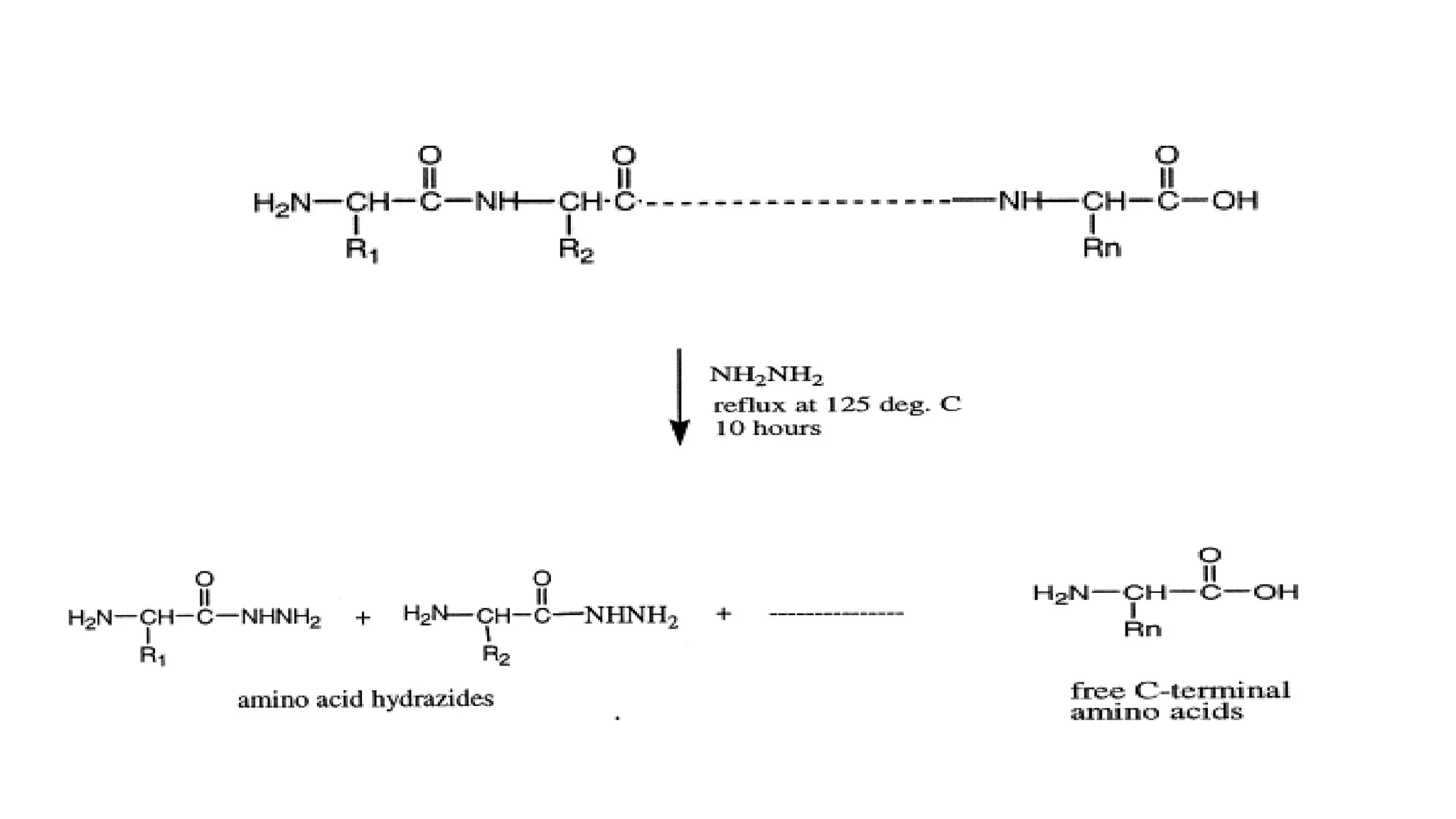
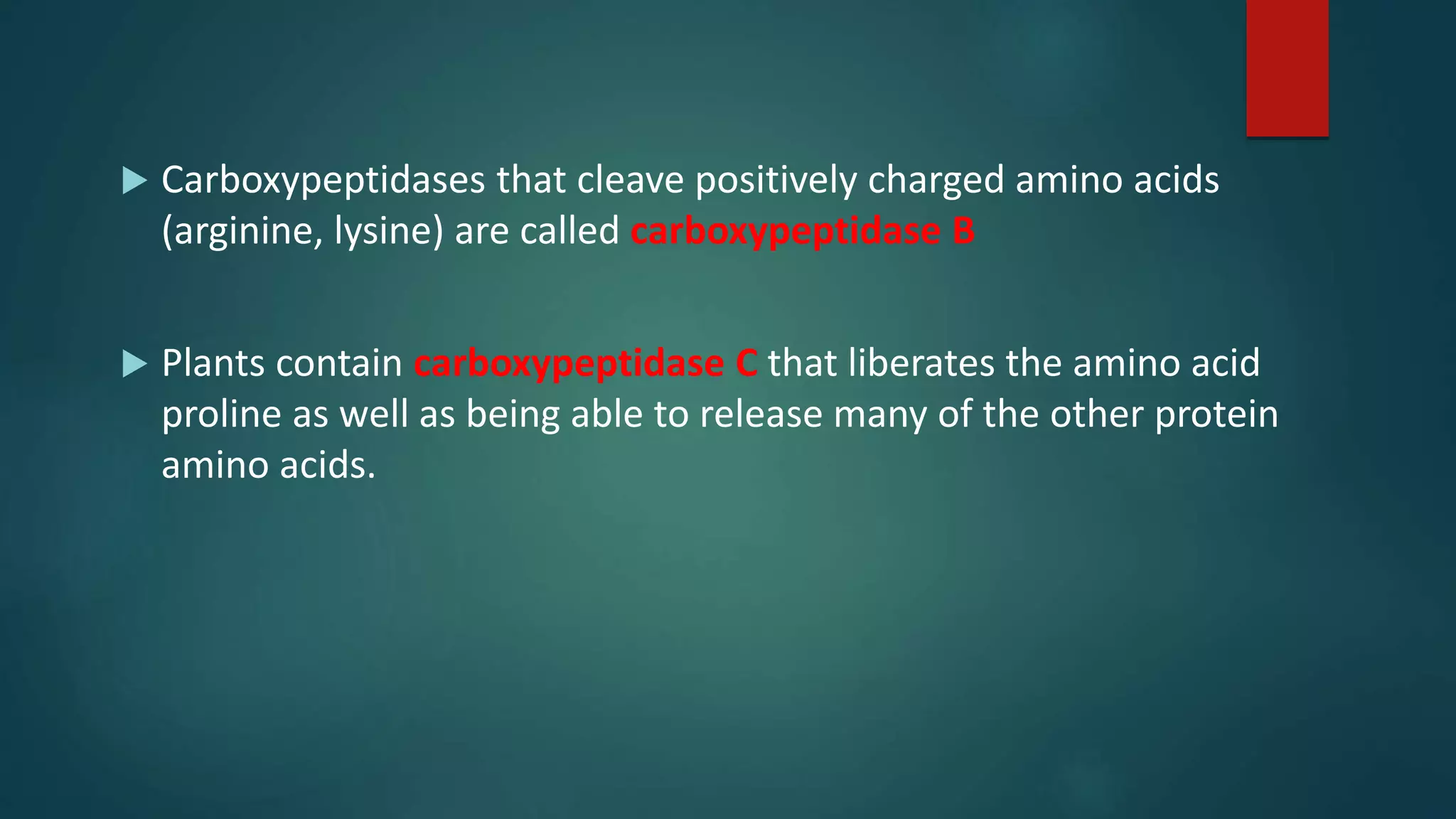
Identification techniques for C-terminus using Akabori method and carboxypeptidases.
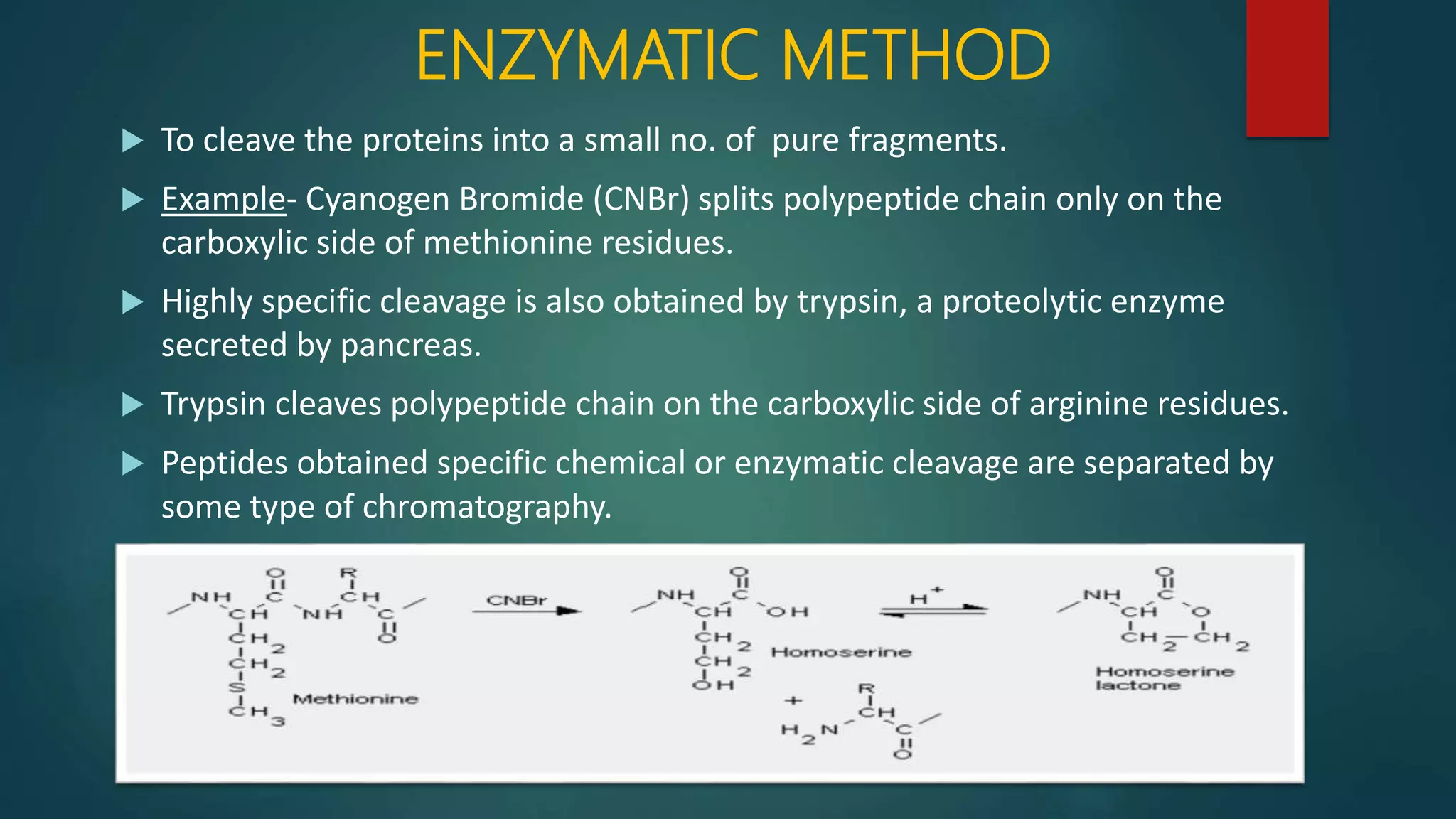
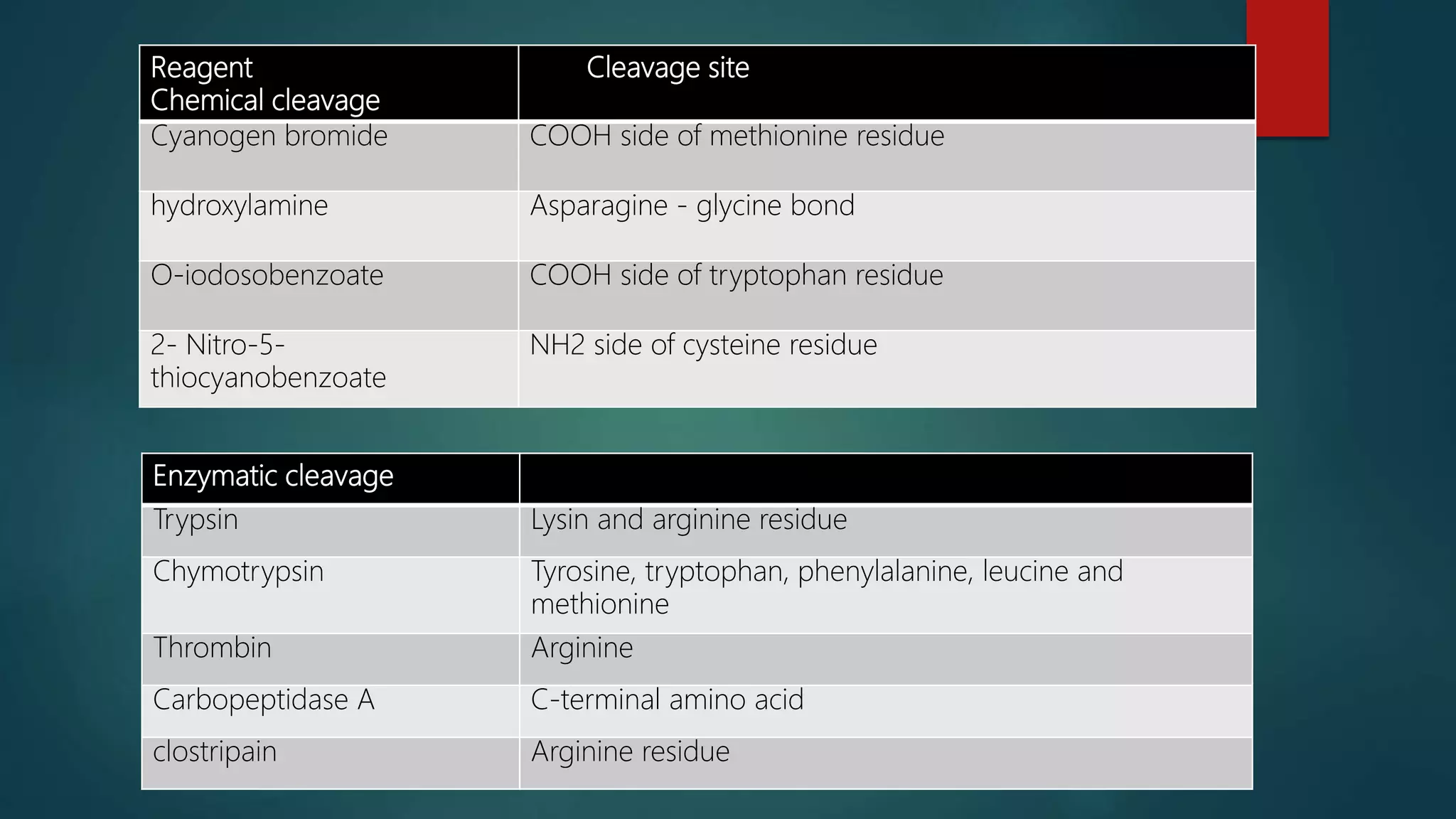
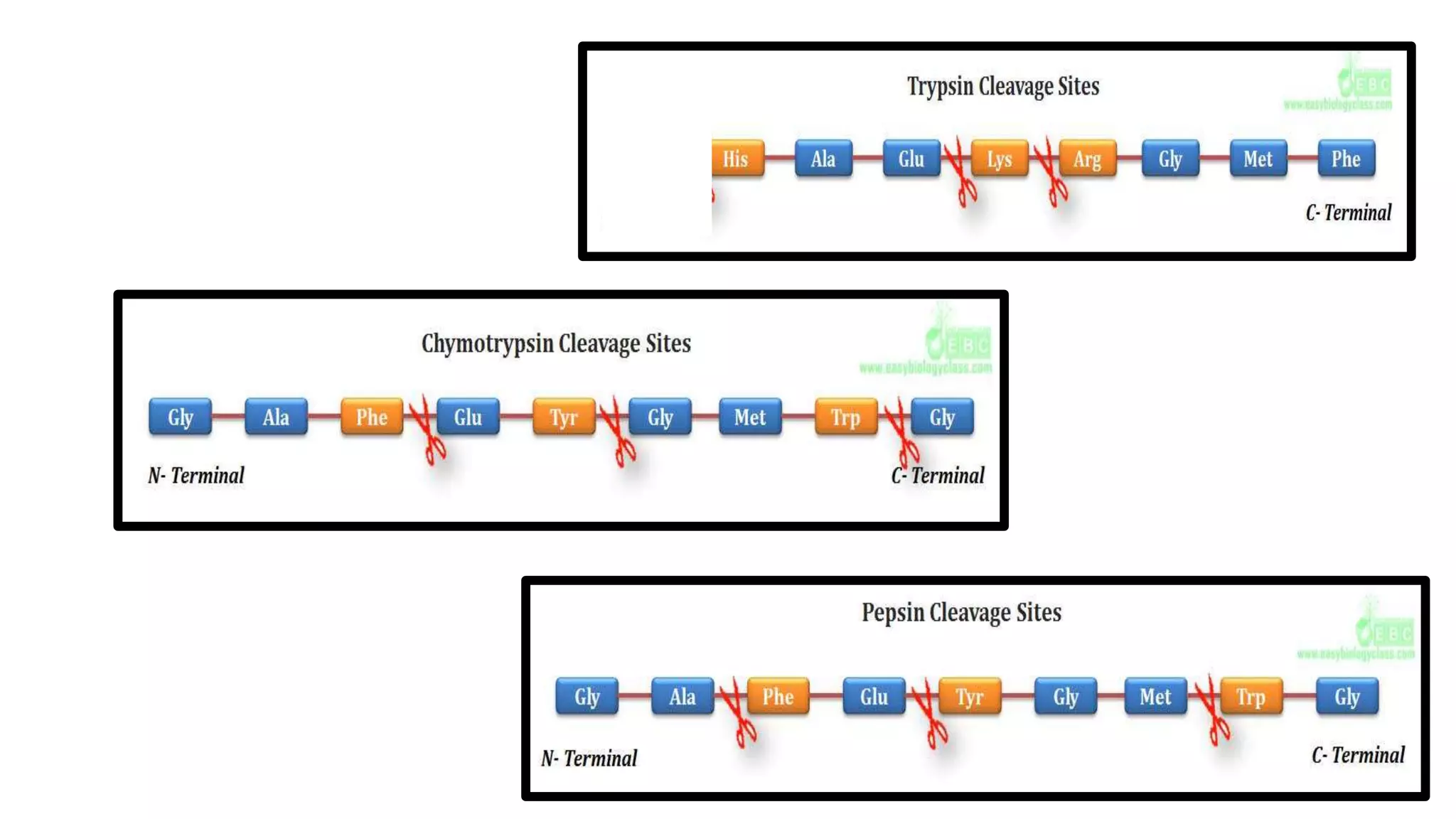
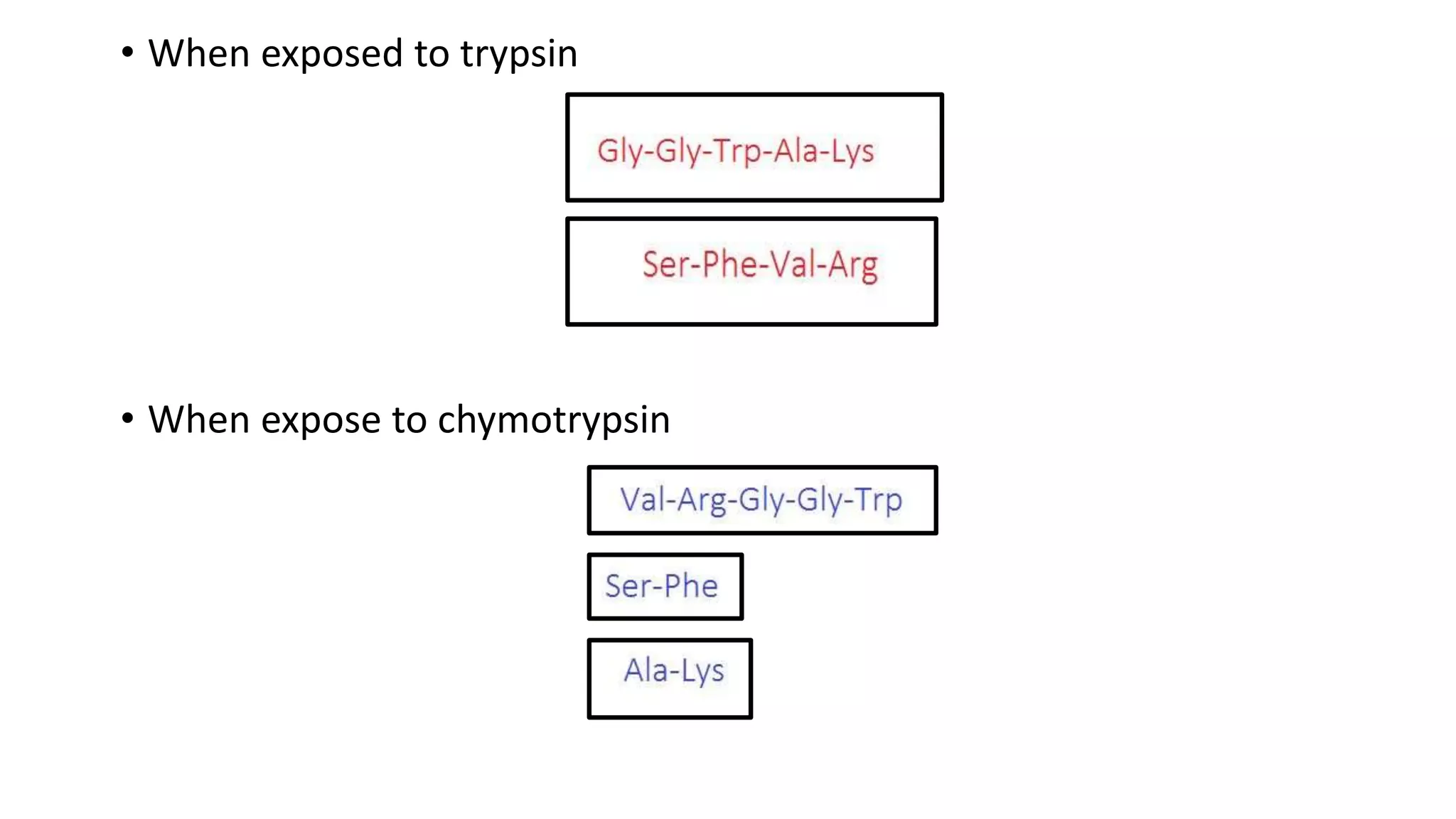
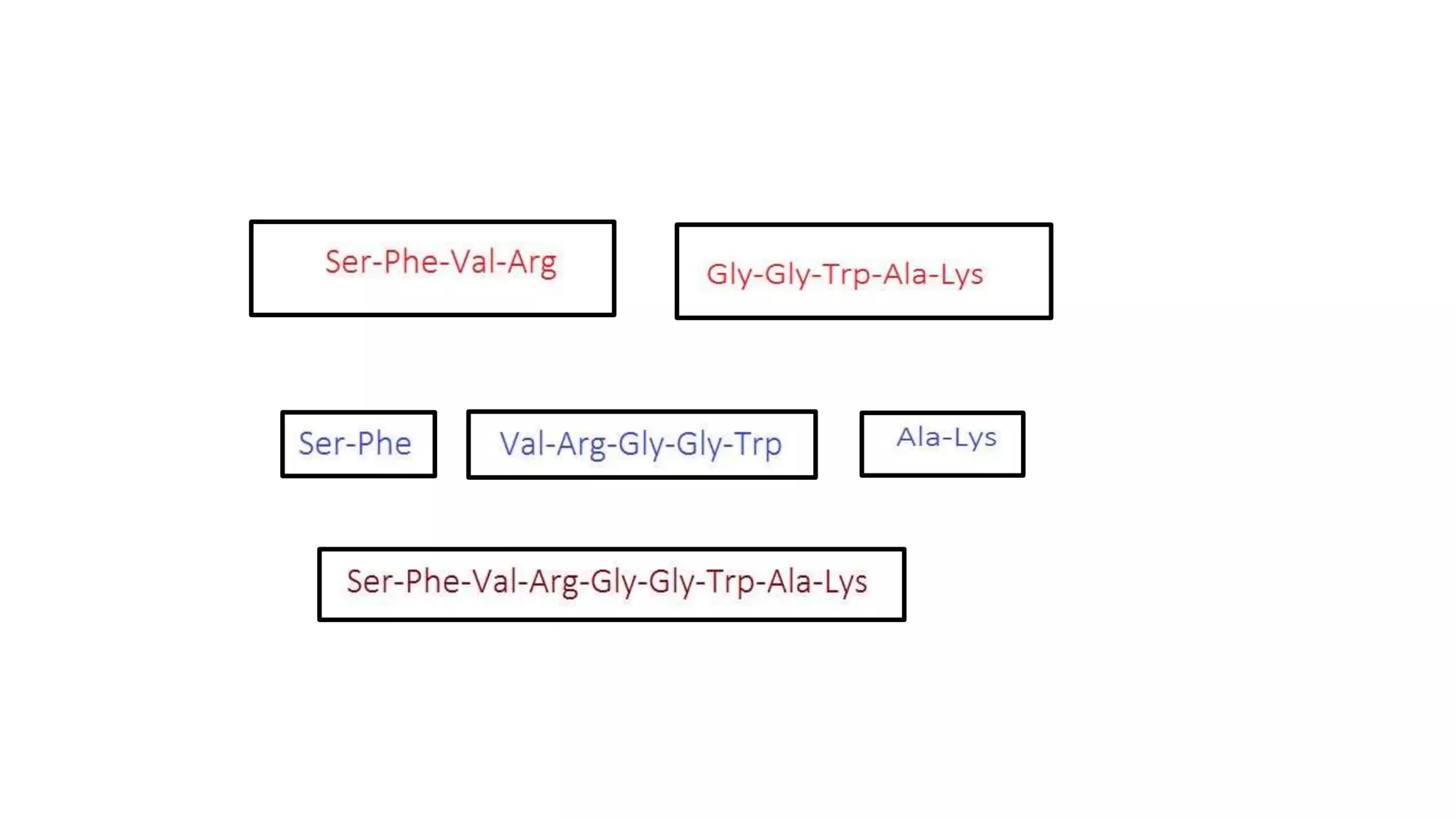
Techniques for cleaving polypeptides into fragments using enzymatic and chemical methods.
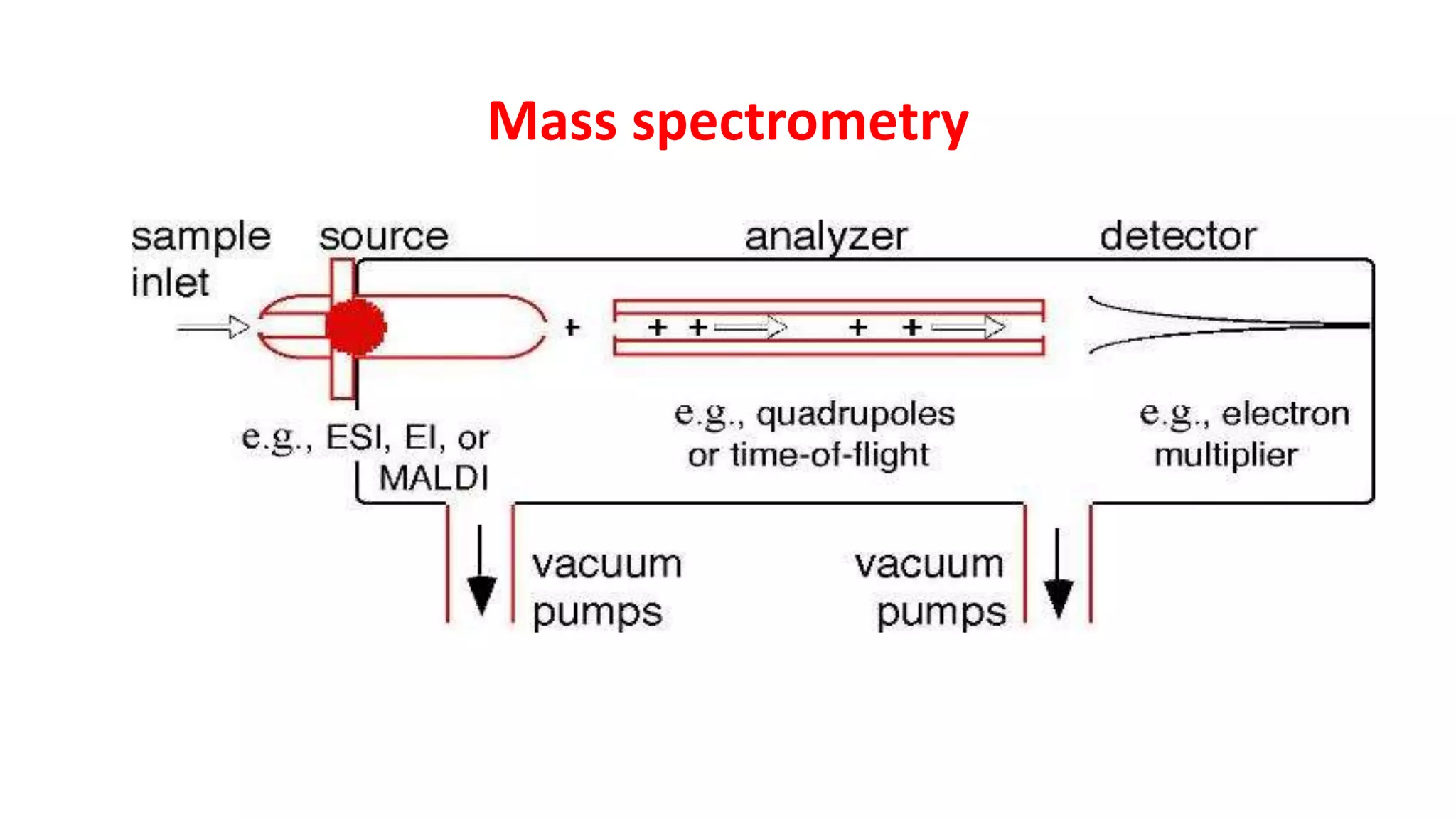
Introduction to mass spectrometry principles and its application in determining molecular weights.
Various ionization methods and their applications in mass spectrometry for analysis of biomolecules.
Advantages of tandem mass spectrometry for detailed structural analysis of complex molecules.
Overview of techniques like ELISA, Western blotting, and immunofluorescence for protein investigation.
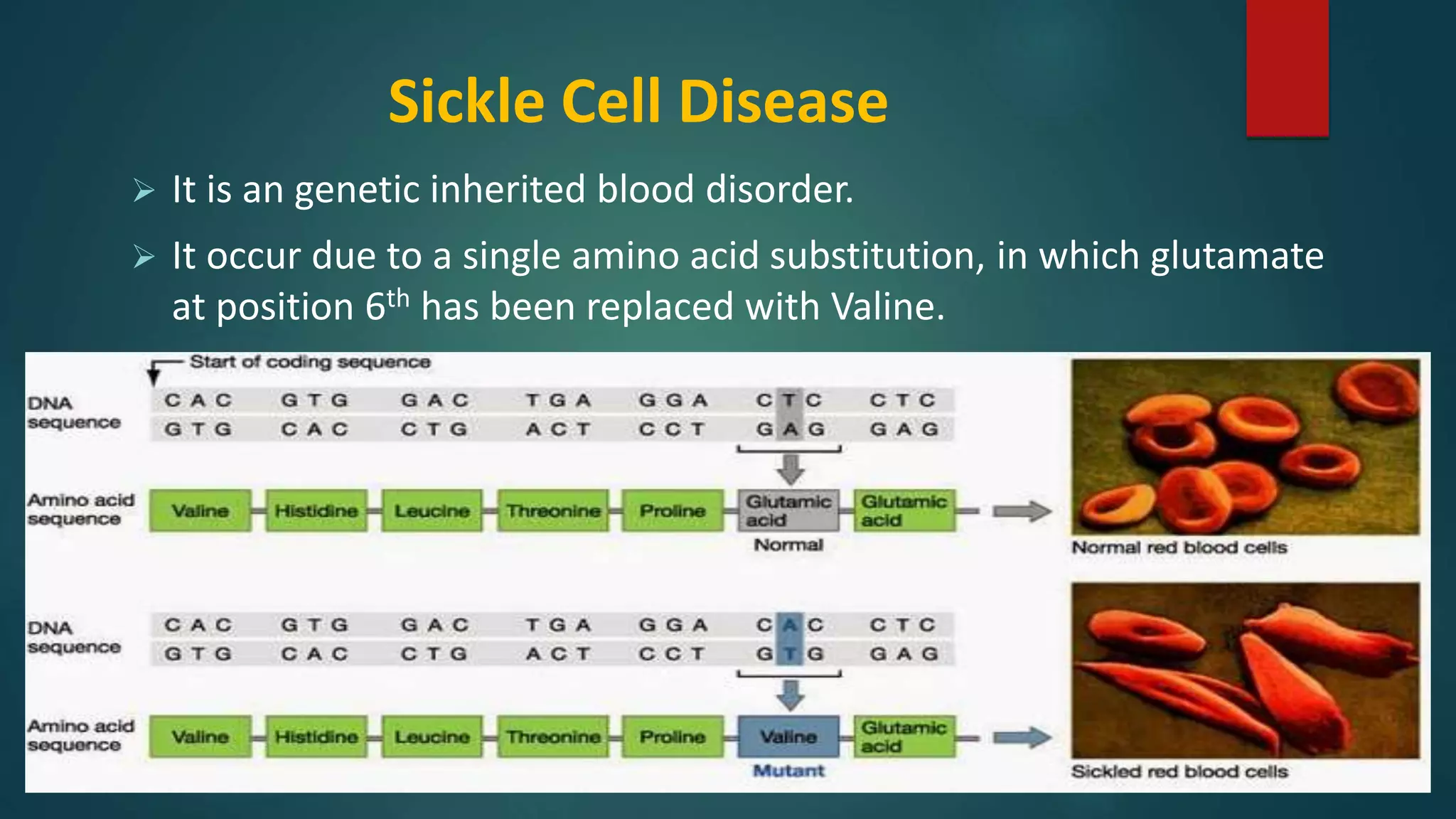
Description of sickle cell disease caused by an amino acid substitution and its physiological effects.























































































