Downloaded 184 times





![ Formation of a secondary structure is the first step in the folding process that a
protein takes to assume its native structure. Characteristic of secondary structure
is the structures known as alpha helices and beta sheets that fold rapidly because
they are stabilized by intramolecular hydrogen bonds, as was first characterized
by Linus Pauling. Formation of intramolecular hydrogen bonds provides another
important contribution to protein stability. Alpha helices are formed by hydrogen
bonding of the backbone to form a spiral shape (refer to figure on the right).[8]
The beta pleated sheet is a structure that forms with the backbone bending over
itself to form the hydrogen bonds (as displayed in the figure to the left). The
hydrogen bonds are between the amide hydrogen and carbonyl carbon of the
peptide bond. There exists anti-parallel beta pleated sheets and parallel beta
pleated sheets where the stability of the hydrogen bonds is stronger in the anti-
parallel beta sheet as it hydrogen bonds with the ideal 180 degree angle
compared to the slanted hydrogen bonds formed by parallel sheets.The alpha
helices and beta pleated sheets can be amphipathic in nature, or contain a
hydrophilic portion and a hydrophobic portion. This property of these secondary
structures aids in the folding of the protein as it aligns the helices and sheets in
such a way where the hydrophilic sides are facing the aqueous environment
surrounding the protein and the hydrophobic sides are facing the hydrophobic
core of the protein. Secondary structure hierarchically gives way to tertiary
structure formation.](https://image.slidesharecdn.com/proteinfolding-170226165229/85/Protein-folding-5-320.jpg)

![ Folding is a spontaneous process independent of energy inputs
from nucleoside triphosphates. The passage of the folded state
is mainly guided by hydrophobic interactions, formation of
intramolecular hydrogen bonds, van der Waals forces, and it is
opposed by conformational entropy.[13] The process of folding
often begins co-translationally, so that the N-terminus of the
protein begins to fold while the C-terminal portion of the protein
is still being synthesized by the ribosome; however, a protein
molecule may fold spontaneously during or after biosynthesis.
While these macromolecules may be regarded as "folding
themselves", the process also depends on the solvent (water or
lipid bilayer),[14] the concentration of salts, the pH, the
temperature, the possible presence of cofactors and of molecular
chaperones. Proteins will have limitations on their folding
abilities by the restricted bending angles or conformations that
are possible. These allowable angles of protein folding are
described with a two-dimensional plot known as the
Ramachandran plot, depicted with psi and phi angles of
allowable rotation](https://image.slidesharecdn.com/proteinfolding-170226165229/85/Protein-folding-7-320.jpg)
![ Protein folding must be thermodynamically favorable within a cell in order for it to be a
spontaneous reaction. Since it is known that protein folding is a spontaneous reaction, then it
must assume a negative Gibbs free energy value. Gibbs free energy in protein folding is
directly related to enthalpy and entropy.[8] For a negative delta G to arise and for protein
folding to become thermodynamically favorable, then either enthalpy, entropy, or both terms
must be favorable.
Entropy is decreased as the water molecules become more orderly near the hydrophobic
solute.
Minimizing the number of hydrophobic side-chains exposed to water is an important driving
force behind the folding process.[16] The hydrophobic effect is the phenomenon in which the
hydrophobic chains of a protein collapse into the core of the protein (away from the
hydrophilic environment).[8] In an aqueous environment, the water molecules tend to
aggregate around the hydrophobic regions or side chains of the protein, creating water shells
of ordered water molecules.[17] An ordering of water molecules around a hydrophobic region
increases order in a system and therefore contributes a negative change in entropy (less
entropy in the system). The water molecules are fixed in these water cages which drives the
hydrophobic collapse, or the inward folding of the hydrophobic groups. The hydrophobic
collapse introduces entropy back to the system via the breaking of the water cages which frees
the ordered water molecules.[8] The multitude of hydrophobic groups interacting within the
core of the globular folded protein contributes a significant amount to protein stability after
folding, because of the vastly accumulated van der Waals forces (specifically London Dispersion
forces).[8] The hydrophobic effect exists as a driving force in thermodynamics only if there is
the presence of an aqueous medium with an amphiphilic molecule containing a large
hydrophobic region.[18] The strength of hydrogen bonds depends on their environment; thus,
H-bonds enveloped in a hydrophobic core contribute more than H-bonds exposed to the
aqueous environment to the stability of the native state.](https://image.slidesharecdn.com/proteinfolding-170226165229/85/Protein-folding-8-320.jpg)
![ Chaperones are a class of proteins that aid in the correct folding of other proteins in vivo.
Chaperones exist in all cellular compartments and interact with the polypeptide chain in order
to allow the native three-dimensional conformation of the protein to form; however,
chaperones themselves are not included in the final structure of the protein they are assisting
in. Chaperones may assist in folding even when the nascent polypeptide is being synthesized
by the ribosome. Molecular chaperones operate by binding to stabilize an otherwise unstable
structure of a protein in its folding pathway, but chaperones do not contain the necessary
information to know the correct native structure of the protein they are aiding; rather,
chaperones work by preventing incorrect folding conformations. In this way, chaperones do
not actually increase the rate of individual steps involved in the folding pathway toward the
native structure; instead, they work by reducing possible unwanted aggregations of the
polypeptide chain that might otherwise slow down the search for the proper intermediate and
they provide a more efficient pathway for the polypeptide chain to assume the correct
conformations.[20] Chaperones are not to be confused with folding catalysts, which actually do
catalyze the otherwise slow steps in the folding pathway. Examples of folding catalysts are
protein disulfide isomerases and peptidyl-prolyl isomerases that may be involved in formation
of disulfide bonds or interconversion between cis and trans stereoisomers, respectively.[21]
Chaperones are shown to be critical in the process of protein folding in vivo because they
provide the protein with the aid needed to assume its proper alignments and conformations
efficiently enough to become "biologically relevant". This means that the polypeptide chain
could theoretically fold into its native structure without the aid of chaperones, as demonstrated
by protein folding experiments conducted in vitro; however, this process proves to be too
inefficient or too slow to exist in biological systems; therefore, chaperones are necessary for
protein folding in vivo. Along with its role in aiding native structure formation, chaperones are
shown to be involved in various roles such as protein transport, degradation, and even allow
denatured proteins exposed to certain external denaturant factors an opportunity to refold into
their correct native structures.](https://image.slidesharecdn.com/proteinfolding-170226165229/85/Protein-folding-9-320.jpg)
![ A fully denatured protein lacks both tertiary and secondary structure, and exists as a so-called
random coil. Under certain conditions some proteins can refold; however, in many cases,
denaturation is irreversible. Cells sometimes protect their proteins against the denaturing
influence of heat with enzymes known as heat shock proteins (a type of chaperone), which
assist other proteins both in folding and in remaining folded. Some proteins never fold in cells
at all except with the assistance of chaperones which either isolate individual proteins so that
their folding is not interrupted by interactions with other proteins or help to unfold misfolded
proteins, allowing them to refold into the correct native structure. This function is crucial to
prevent the risk of precipitation into insoluble amorphous aggregates. The external factors
involved in protein denaturation or disruption of the native state include temperature, external
fields (electric, magnetic),[26] molecular crowding, and even the limitation of space, which can
have a big influence on the folding of proteins. High concentrations of solutes, extremes of pH,
mechanical forces, and the presence of chemical denaturants can contribute to protein
denaturation, as well. These individual factors are categorized together as stresses.
Chaperones are shown to exist in increasing concentrations during times of cellular stress and
help the proper folding of emerging proteins as well as denatured or misfolded ones.
Under some conditions proteins will not fold into their biochemically functional forms.
Temperatures above or below the range that cells tend to live in will cause thermally unstable
proteins to unfold or denature (this is why boiling makes an egg white turn opaque). Protein
thermal stability is far from constant, however; for example, hyperthermophilic bacteria have
been found that grow at temperatures as high as 122 °C,which of course requires that their full
complement of vital proteins and protein assemblies be stable at that temperature or above.](https://image.slidesharecdn.com/proteinfolding-170226165229/85/Protein-folding-10-320.jpg)

![ After synthesis, proteins typically fold into a particular three-dimensional conformation that is
the most thermodynamically favorable: their native state.[5] This folding process is driven by
the hydrophobic effect: a tendency for hydrophobic (water-fearing) portions of the protein to
shield itself from the hydrophilic (water-loving) environment of the cell by burying into the
interior of the protein. Thus, the exterior of a protein is typically hydrophilic, whereas the
interior is typically hydrophobic.
Protein structures are stabilized by non-covalent interactions and disulfide bonds between two
cysteine residues. The non-covalent interactions include ionic interactions and weak van der
waals interactions. Ionic interactions form between an anion and a cation and form salt bridges
that help stabilize the protein. Van der waals interactions include nonpolar interactions (i.e.
London dispersion forces) and polar interactions (i.e. hydrogen bonds, dipole-dipole bond).
These play an important role in a protein's secondary structure, such as forming an alpha helix
or a beta sheet, and tertiary structure. Interactions between amino acid residues in a specific
protein are very important in that protein's final structure.
When there are changes in the non-covalent interactions, as may happen with a change in the
amino acid sequence, the protein is susceptible to misfolding or unfolding. In these cases, if
the cell does not assist the protein in re-folding, or degrade the unfolded protein, the
unfolded/misfolded protein may aggregate, in which the exposed hydrophobic portions of the
protein may interact with the exposed hydrophobic patches of other proteins.[6][7] There are
three main types of protein aggregates that may form: amorphous aggregates, oligomers, and
amyloid fibrils](https://image.slidesharecdn.com/proteinfolding-170226165229/85/Protein-folding-12-320.jpg)
![ Protein aggregation can occur due to a variety of causes. There are four classes that
these causes can be categorized into, which are detailed below.
Mutations[edit]
Mutations that occur in the DNA sequence may or may not affect the amino acid
sequence of the protein. When the sequence is affected, a different amino acid may
change the interactions between the side chains that affect the folding of the protein.
This can lead to exposed hydrophobic regions of the protein that aggregate with the
same misfolded/unfolded protein or a different protein.
In addition to mutations in the affected proteins themselves, protein aggregation could
also be caused indirectly through mutations in proteins in regulatory pathways such as
the refolding pathway (molecular chaperones) or the ubiquitin-proteasome pathway
(ubiquitin ligases).[9] Chaperones help with protein refolding by providing a safe
environment for the protein to fold. Ubiquitin ligases target proteins for degradation
through ubiquitin modification.
Problems with protein synthesis[edit]
Protein aggregation can be caused by problems that occur during transcription or
translation. During transcription, DNA is copied into mRNA, forming a strand of pre-
mRNA that undergoes RNA processing to form mRNA.[10] During translation, ribosomes
and tRNA help translate the mRNA sequence into an amino acid sequence.[10] If
problems arise during either step, making an incorrect mRNA strand and/or an incorrect
amino acid sequence, this can cause the protein to misfold, leading to protein
aggregation.](https://image.slidesharecdn.com/proteinfolding-170226165229/85/Protein-folding-13-320.jpg)
![ Environmental stresses
Environmental stresses such as extreme temperatures and pH or oxidative stress can also lead to protein
aggregation.[11] One such disease is cryoglobulinemia.
Extreme temperatures can weaken and destabilize the non-covalent interactions between the amino acid
residues. pHs outside of the protein's pH range can change the protonation state of the amino acids, which
can increase or decrease the non-covalent interactions. This can also lead to less stable interactions and
result in protein unfolding.
Oxidative stress can be caused by radicals such as reactive oxygen species (ROS). These unstable radicals
can attack the amino acid residues, leading to oxidation of side chains (e.g. aromatic side chains,
methionine side chains) and/or cleavage of the polypeptide bonds.[12] This can affect the non-covalent
interactions that hold the protein together correctly, which can cause protein destabilization, and may
cause the protein to unfold.
Aging
Cells have mechanisms that can refold or degrade protein aggregates. However, as cells age, these control
mechanisms are weakened and the cell is less able to resolve the aggregates.
The hypothesis that protein aggregation is a causative process in aging is testable now since some models
of delayed aging are in hand. If the development of protein aggregates was an aging independent process,
slowing down aging will show no effect on the rate of proteotoxicity over time. However, if aging is
associated with decline in the activity of protective mechanisms against proteotoxicity, the slow aging
models would show reduced aggregation and proteotoxicity. To address this problem several toxicity
assays have been done in C. elegans. These studies indicated that reducing the activity of insulin/IGF
signaling (IIS), a prominent aging regulatory pathway protects from neurodegeneration-linked toxic
protein aggregation. The validity of this approach has been tested and confirmed in mammals as reducing
the activity of the IGF-1 signaling pathway protected Alzheimer's model mice from the behavioral and
biochemical impairments associated with the disease.](https://image.slidesharecdn.com/proteinfolding-170226165229/85/Protein-folding-14-320.jpg)
![ There are two main protein quality control systems in the cell that are responsible
for eliminating protein aggregates. Misfolded proteins can get refolded by the bi-
chaperone system or degraded by the ubiquitin proteasome system or autophagy.
Refolding[edit]
The bi-chaperone system utilizes the Hsp70 (DnaK-DnaJ-GrpE in E. coli and Ssa1-
Ydj1/Sis1-Sse1/Fe1 in yeast) and Hsp100 (ClpB in E. coli and Hsp104 in yeast)
chaperones for protein disaggregation and refolding.
Hsp70 interacts with the protein aggregates and recruits Hsp100. Hsp70 stabilizes
an activated Hsp100. Hsp100 proteins have aromatic pore loops that are used for
threading activity to disentangle single polypeptides. This threading activity can
be initiated at the N-terminus, C-terminus or in the middle of the polypeptide.
The polypeptide gets translocated through Hsp100 in a series of steps, utilizing
an ATP at each step. The polypeptide unfolds and is then allowed to refold either
by itself or with the help of heat shock proteins.
Degradation[edit]
Misfolded proteins can be eliminated through the ubiquitin-proteasome system
(UPS). This consists of an E1-E2-E3 pathway that ubiquinates proteins to mark
them for degradation. In eukaryotes, the proteins get degraded by the 26S
proteasome. In mammalian cells, the E3 ligase, carboxy-terminal Hsp70
interacting protein (CHIP), targets Hsp70-bound proteins. In yeast, the E3 ligases
Doa10 and Hrd1 have similar functions on endoplasmic reticulum proteins.](https://image.slidesharecdn.com/proteinfolding-170226165229/85/Protein-folding-15-320.jpg)






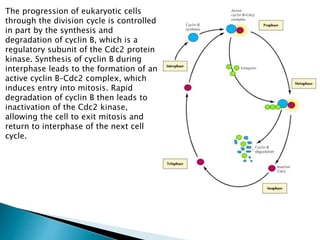



The document discusses protein folding, which is the process by which a polypeptide chain folds into its characteristic and functional three-dimensional structure. It describes the four levels of protein structure: primary, secondary, tertiary, and quaternary. Key drivers of folding are the hydrophobic effect and formation of hydrogen bonds. Chaperone proteins assist in protein folding in vivo. Factors such as mutations, errors in synthesis, environmental stresses, and aging can cause proteins to misfold and aggregate, which is associated with various diseases. Cells use molecular chaperones and protein degradation systems to prevent aggregation, but these become less effective with age.













































































![Polymer [ बहुलक ] Chemistry Notes PDF - Irfanullah Mehar - JJ Sir Chemistry.pdf](https://cdn.slidesharecdn.com/ss_thumbnails/polymerchemistrynotespdf-irfanullahmehar-jjsirchemistry-260210172118-3f9b37f7-thumbnail.jpg?width=640&height=640&fit=bounds)









