Downloaded 165 times




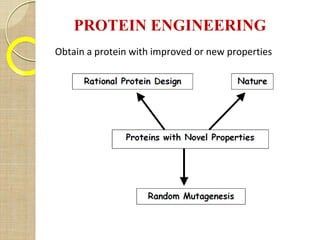
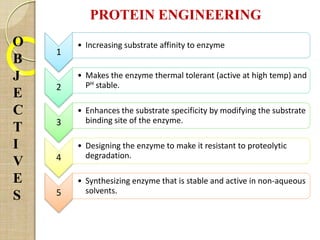








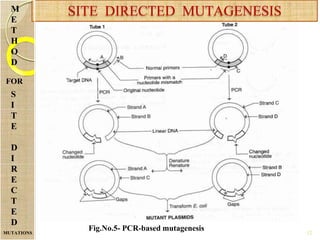









The document discusses protein engineering and techniques used for it. Protein engineering involves altering cloned DNA to modify protein properties. It merges molecular biology, protein chemistry, and other disciplines. Techniques include genetic modifications like site-directed mutagenesis and chemical modifications. Site-directed mutagenesis allows specific changes to the DNA base using methods like oligonucleotide primers and PCR. This allows investigation of protein function and commercial applications like creating detergent-stable enzymes. Protein engineering has applications in increasing stability, activity and investigating protein properties.






















































































