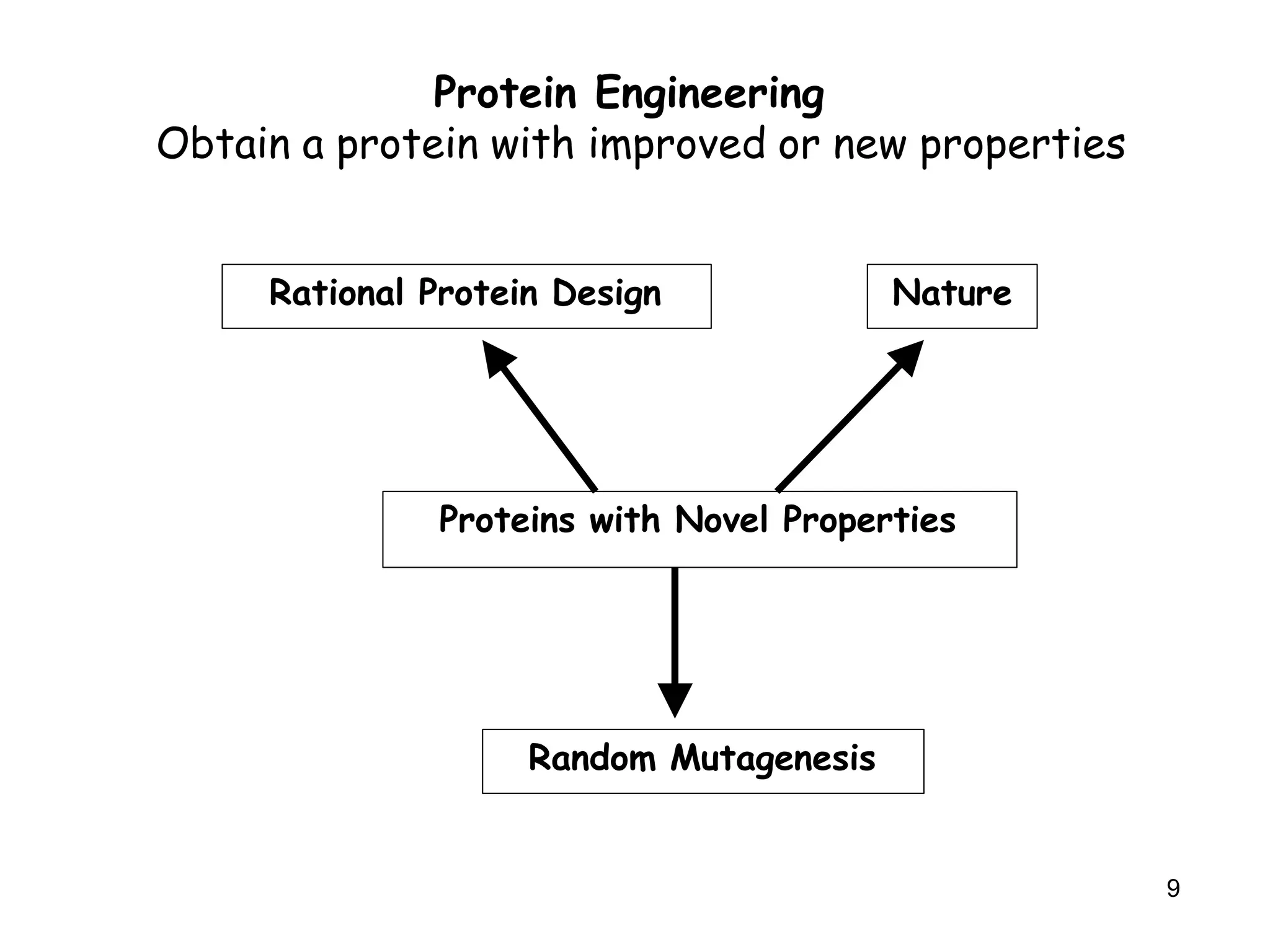
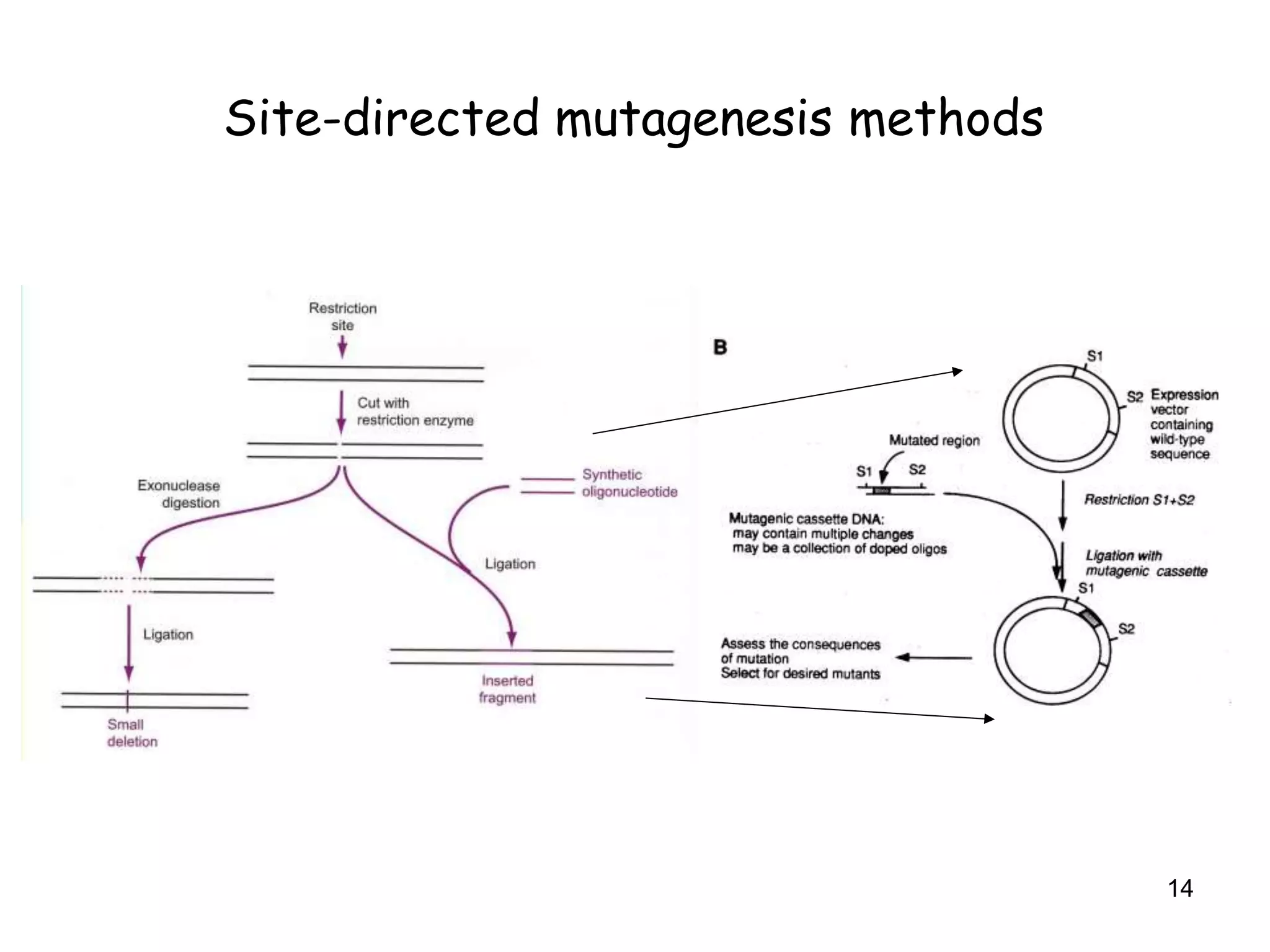
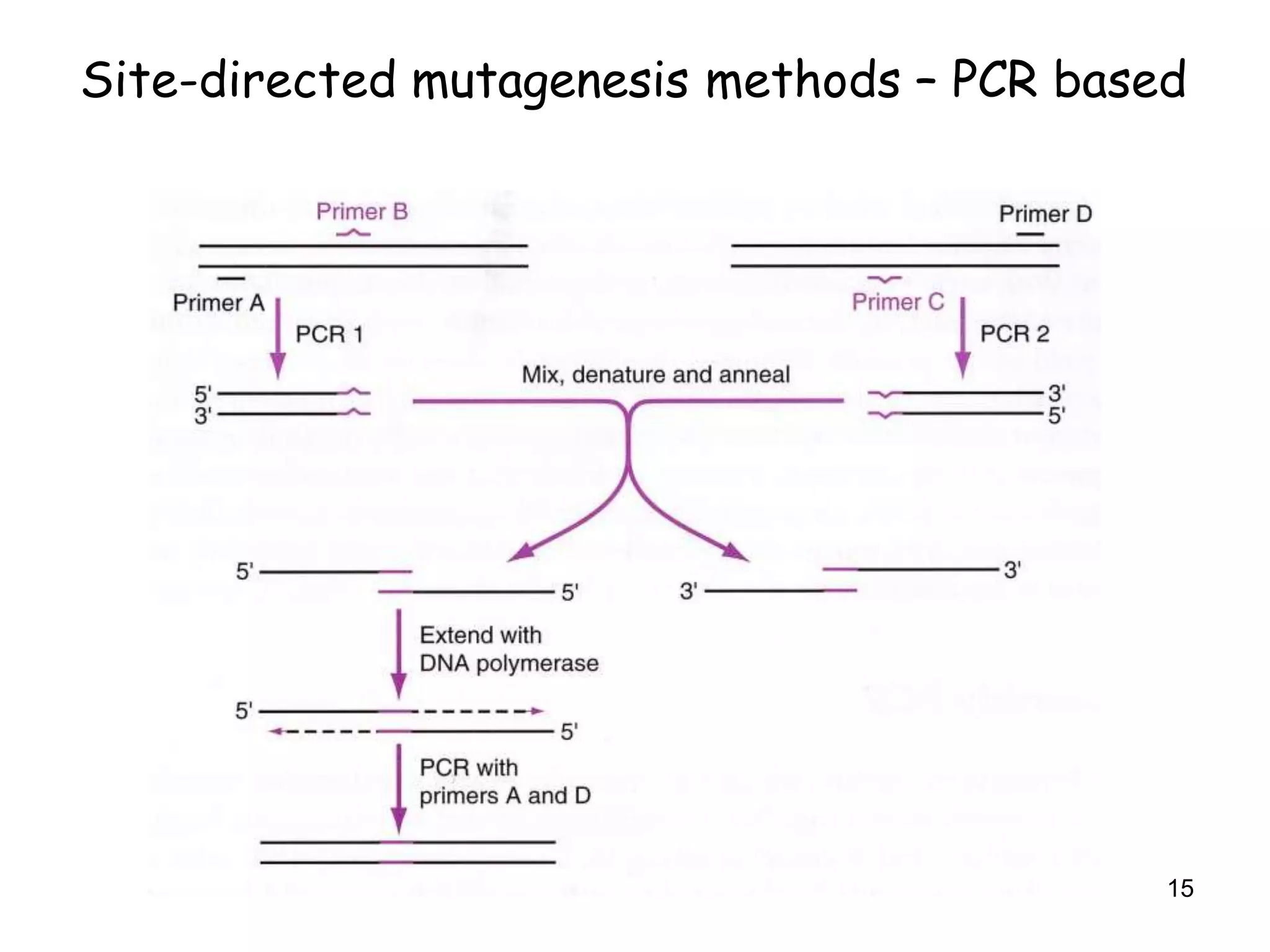
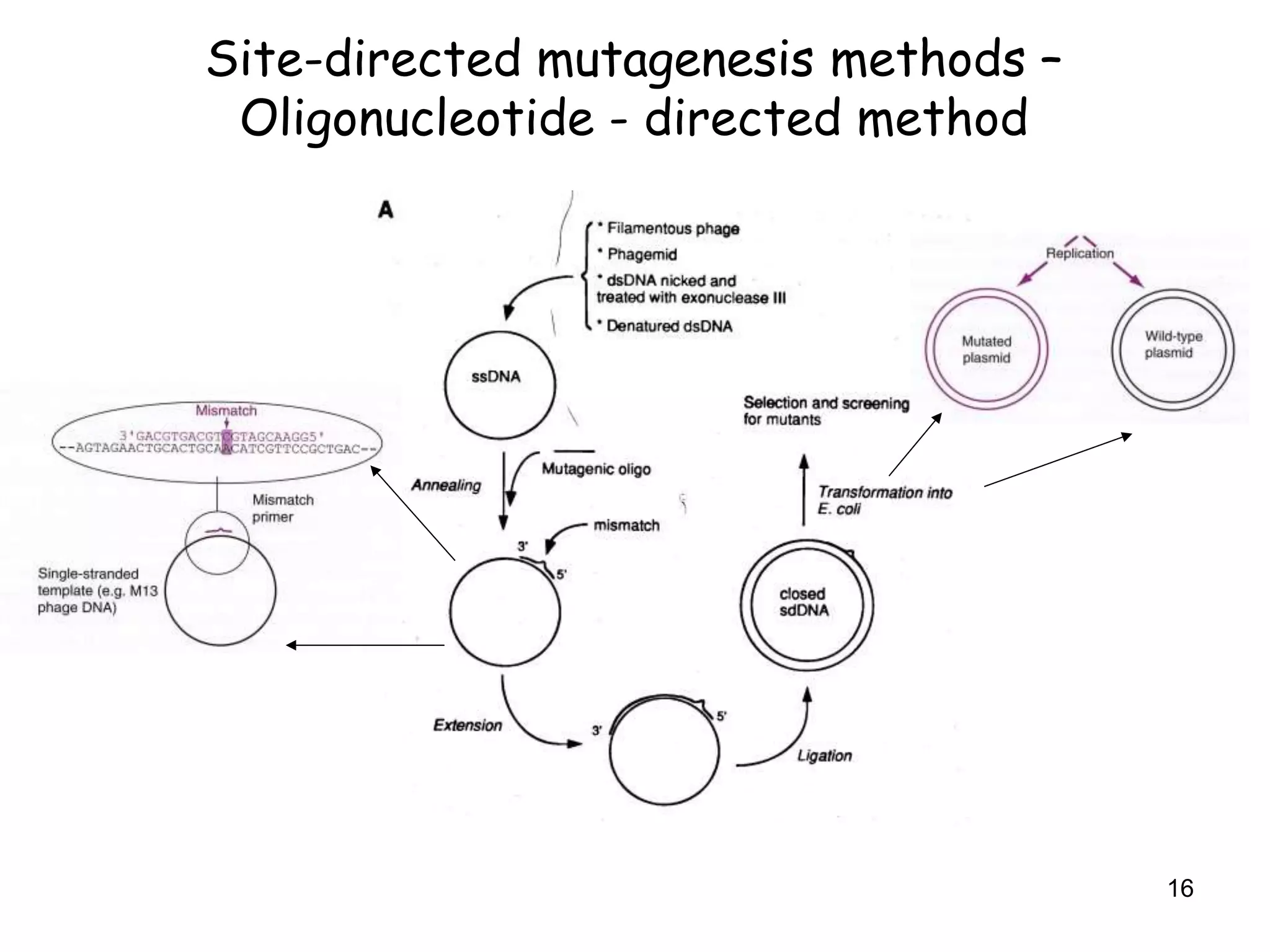
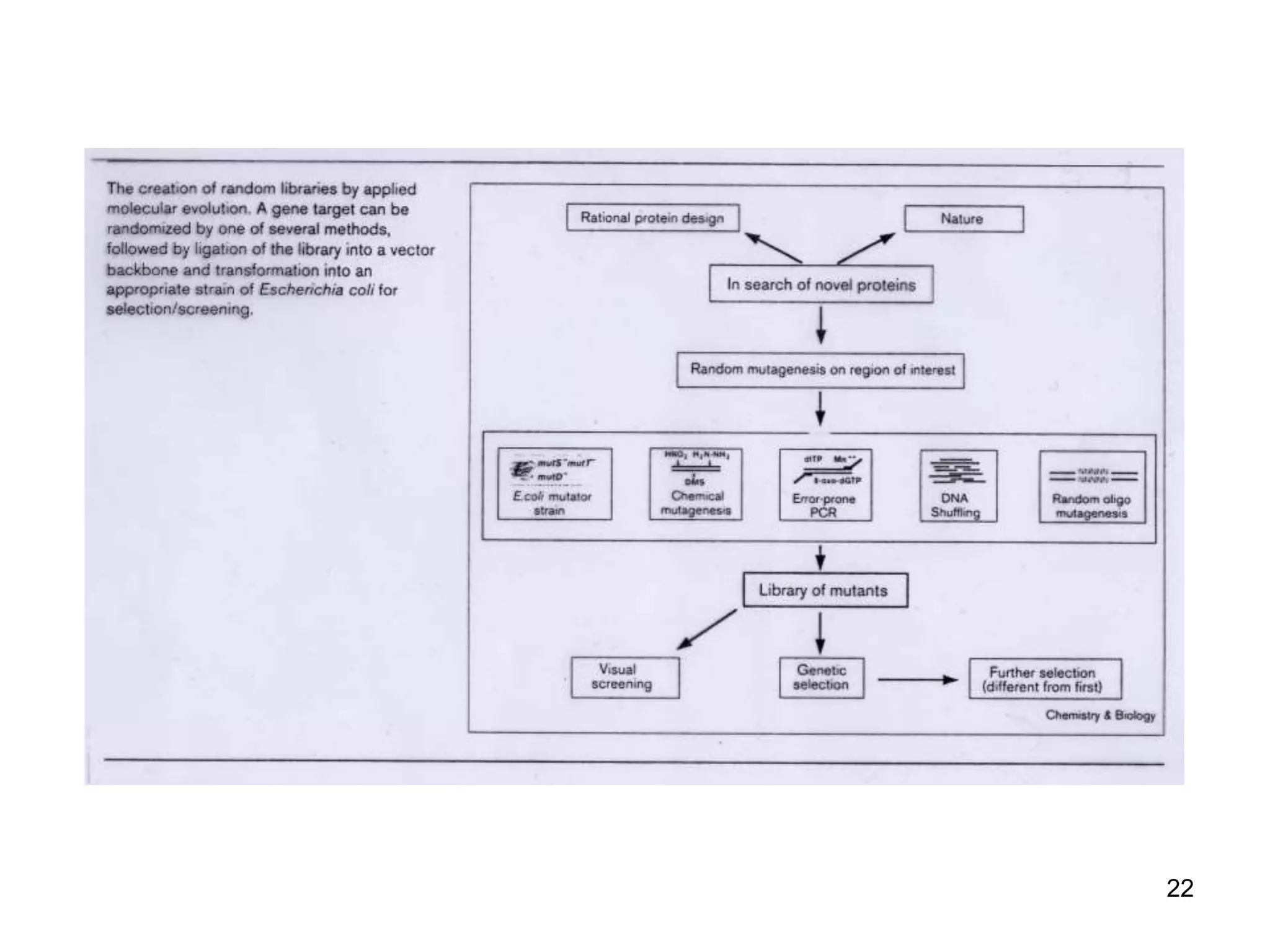
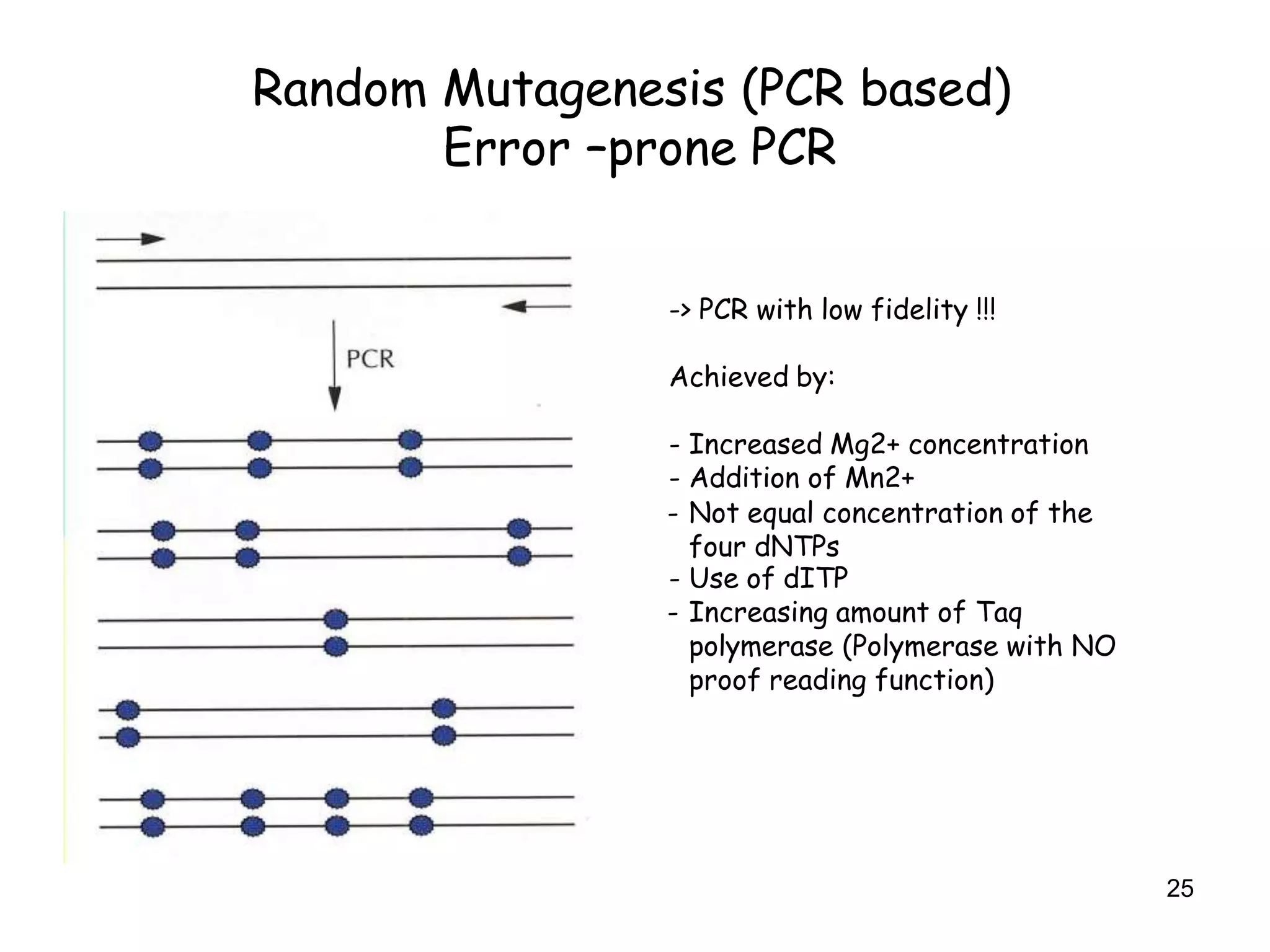
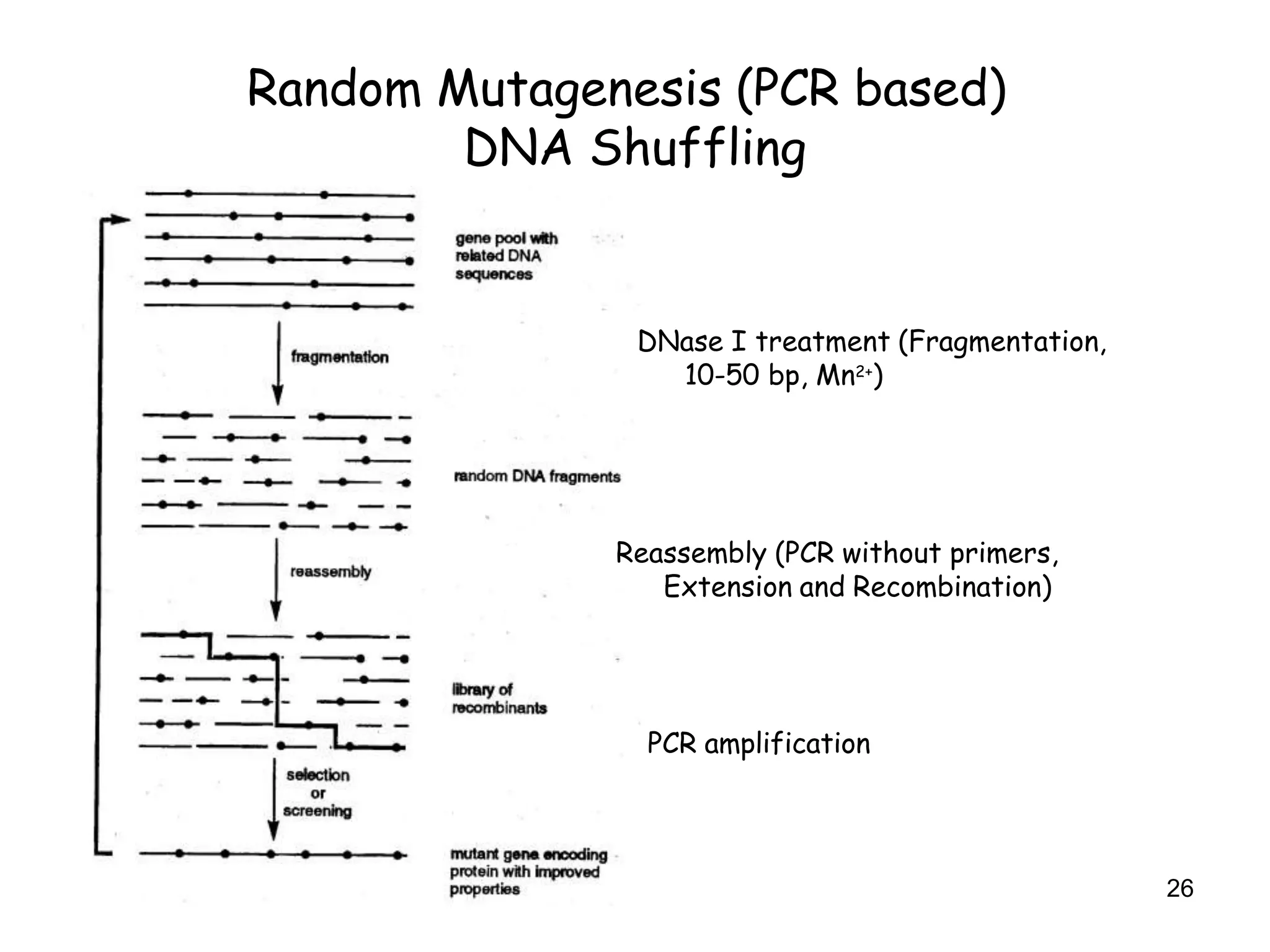
The document discusses protein engineering, focusing on methods like rational design, directed evolution, and peptidomimetics, which aim to modify protein structures for enhanced functions. Key objectives include creating superior enzymes, producing biological compounds, and ensuring enzymes can operate under industrial conditions. Additionally, it covers flow cytometry as a technique for analyzing proteins and cellular characteristics.