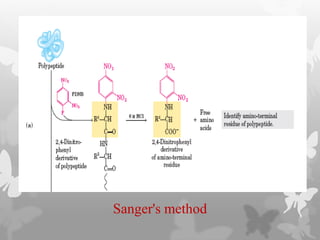
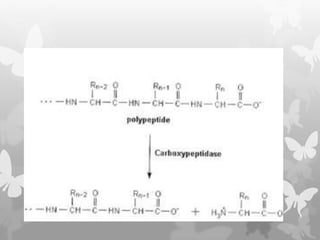
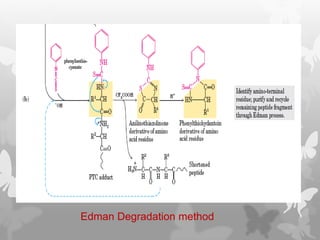
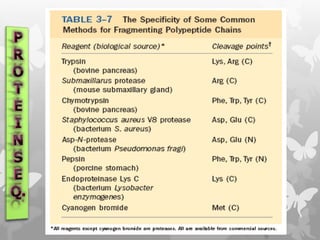
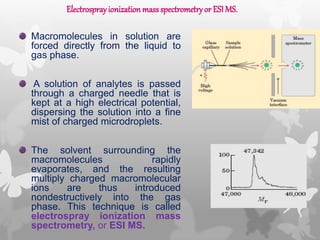
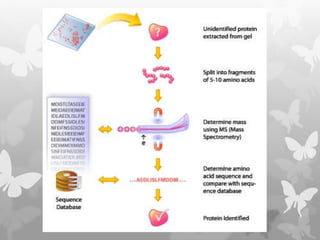
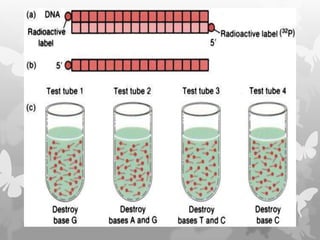
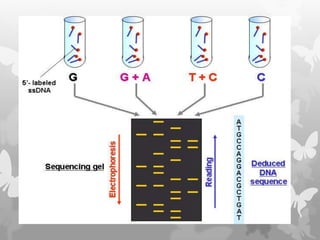
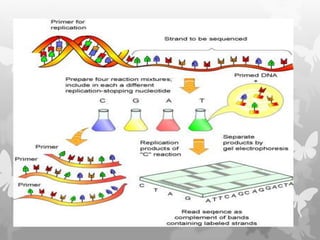
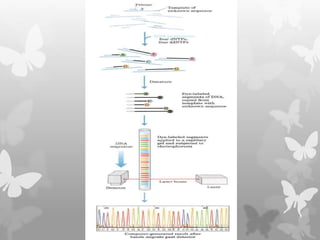
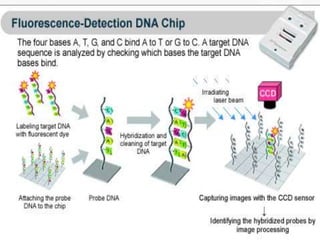
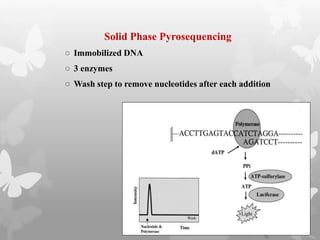
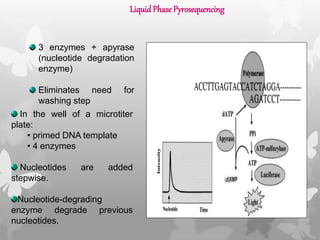
This document provides an extensive overview of protein and nucleic acid sequencing, detailing techniques, history, and methodologies, including Edman degradation and mass spectrometry. It covers the essential steps for determining amino acid and nucleotide sequences, as well as the evolution and applications of sequencing technologies. With references to significant advancements and institutions involved, it highlights the importance of these techniques in biological research and their role in various fields.