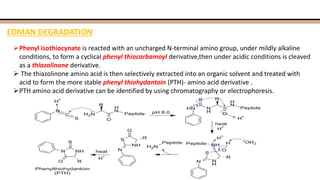
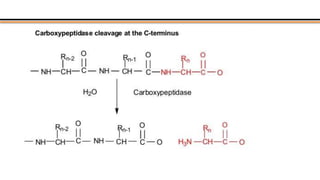
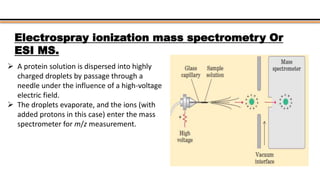
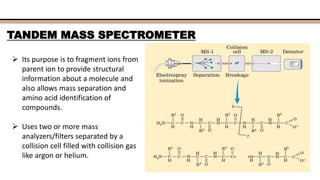
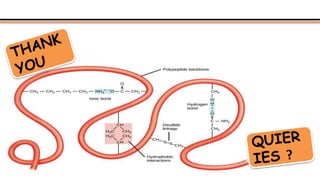
The document discusses protein sequencing, a method for determining the amino acid sequence of proteins, detailing historical milestones and various sequencing techniques such as mass spectrometry and Edman degradation. It emphasizes the importance of amino acid composition before sequencing and highlights the applications of protein sequencing in biological and medical research. Additionally, it references key literature and online resources related to the topic.

































































































![谷歌留痕技术 [ 𝙩𝙤𝙥 𝟮𝟯𝟯. 𝙘 𝙤𝙢 ]](https://cdn.slidesharecdn.com/ss_thumbnails/top233-260130174328-3833018c-thumbnail.jpg?width=640&height=640&fit=bounds)



