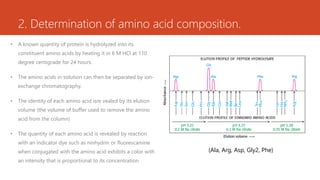
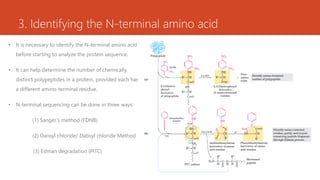
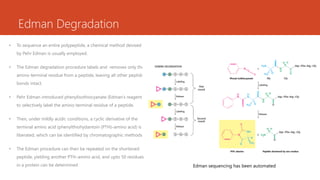
This document discusses several methods for DNA and protein sequencing. It describes automated DNA sequencing which is based on the Sanger method but uses fluorescent labels and allows direct computer storage of sequence data. It then discusses various methods for protein sequencing including purification, amino acid composition analysis, N-terminal sequencing using Edman degradation or other methods, C-terminal sequencing, breaking disulfide bonds, cleaving the protein into peptides, ordering peptides by overlap, and locating disulfide bonds. Newer methods discussed are using genomic data and mass spectrometry techniques.