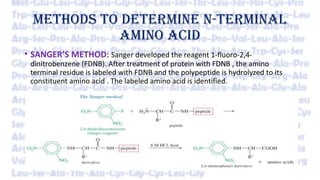
Amino acid sequencing determines the order of amino acids in a protein. Frederick Sanger determined the first protein sequence in 1953 using N-terminal analysis methods like Edman degradation. Large proteins are sequenced by first breaking them into smaller fragments using enzymes or chemicals, then determining the sequence of individual fragments and combining sequences to deduce the full protein sequence. Modern techniques like mass spectrometry have made sequencing faster and applicable to modified proteins.