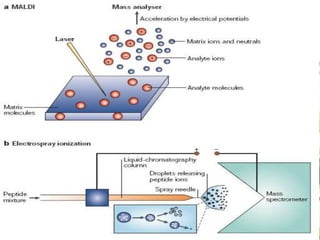
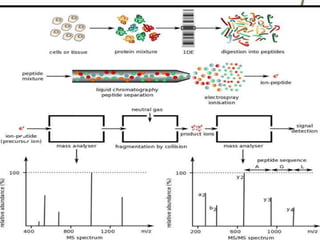
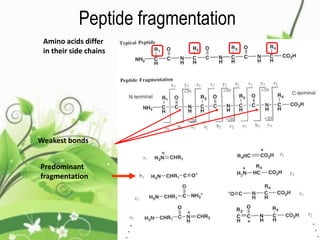
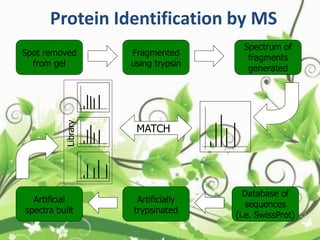
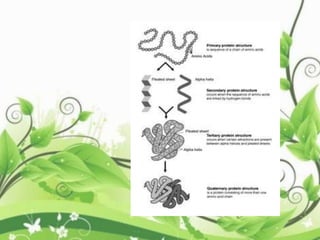
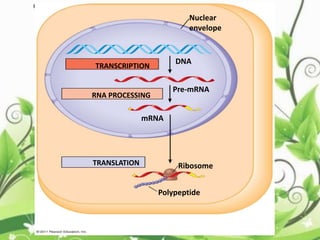
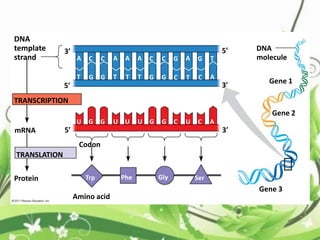
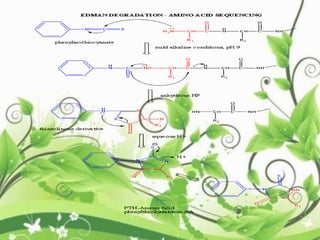
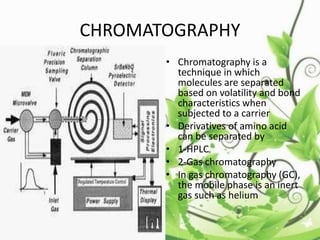
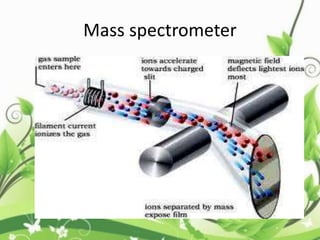
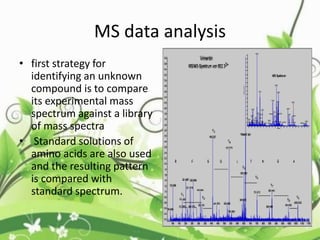
Protein sequencing determines the order of amino acids in a protein. The two major methods are Edman degradation and mass spectrometry. Edman degradation involves labeling the N-terminal amino acid, cleaving it off, and identifying it, repeating the process one amino acid at a time to deduce the sequence. Mass spectrometry involves ionizing and separating protein fragments by mass/charge ratio to determine sequence. Protein sequencing is useful for identifying unknown proteins and characterizing modifications.

















![• Throughout his year at Princeton, Edman was able to
conduct enough experiments to understand that it was
feasible to use reagents like FDNB and PITC to determine
amino acid sequence.
• Edman returned to Sweden in 1947 and after two more
years of work he was able to publish his paper that would
describe the first successful method to sequence proteins
[1]
• This ground breaking paper described a method to
determine the amino acid sequence of a protein and would
come to be known as the Edman Degradation.](https://image.slidesharecdn.com/proteinsequencing-170731104133/85/Protein-sequencing-19-320.jpg)


















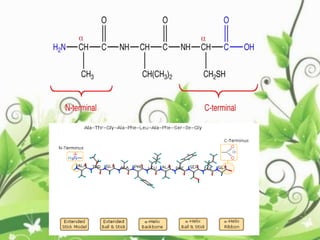


![C-terminal domain:
• The C-terminal domain of some proteins has
specialized functions. In humans, the CTD
of RNA polymerase II typically consists of up to
52 repeats of the sequence Tyr-Ser-Pro-Thr-
Ser-Pro-Ser.[1] This allows other proteins to
bind to the C-terminal domain of RNA
polymerase in order to activate polymerase
activity. These domains then involved in
the initiation of DNA transcription.](https://image.slidesharecdn.com/proteinsequencing-170731104133/85/Protein-sequencing-47-320.jpg)