Downloaded 406 times



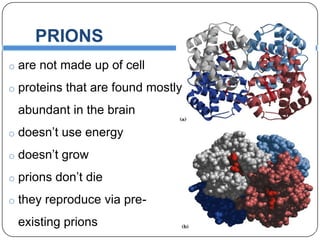


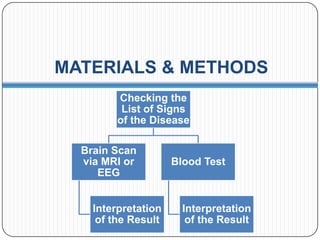

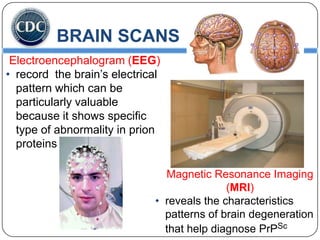
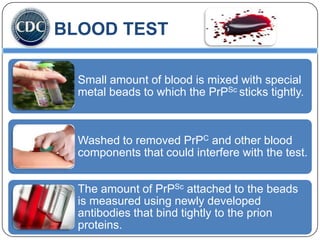
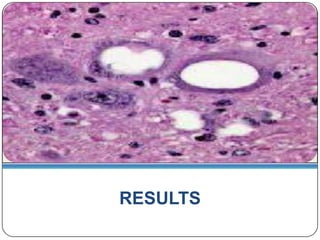
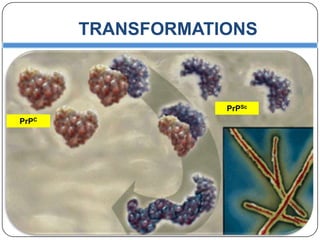
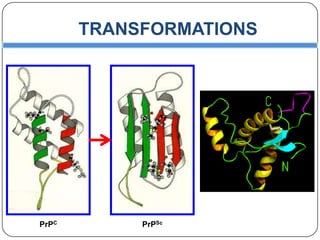
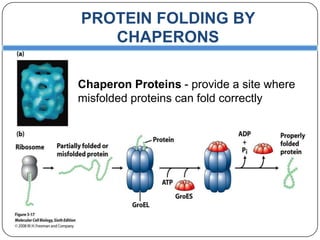
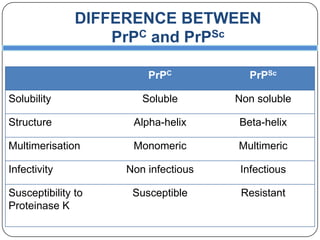
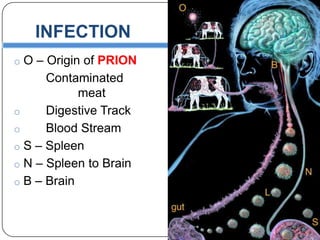
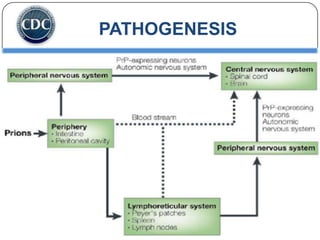
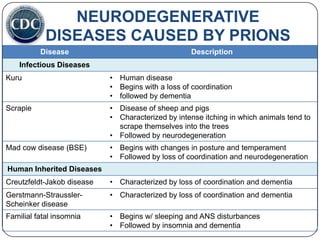
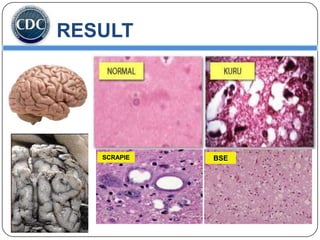
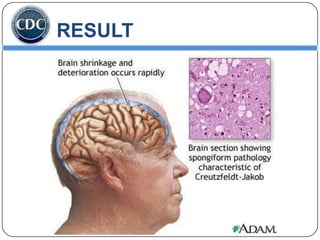




Prions are infectious proteins responsible for neurodegenerative diseases in humans and animals, with early signs including depression and coordination loss. Diagnosis may involve brain scans and blood tests to detect abnormal prion proteins, with diseases like kuru and mad cow disease noted. The study emphasizes the importance of proper meat cooking and awareness of early symptoms for effective intervention.












































![HUMAN_PRION_DISEASES[ A component in virology1].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/humanpriondiseases1-240611104922-95f81163-thumbnail.jpg?width=640&height=640&fit=bounds)

































