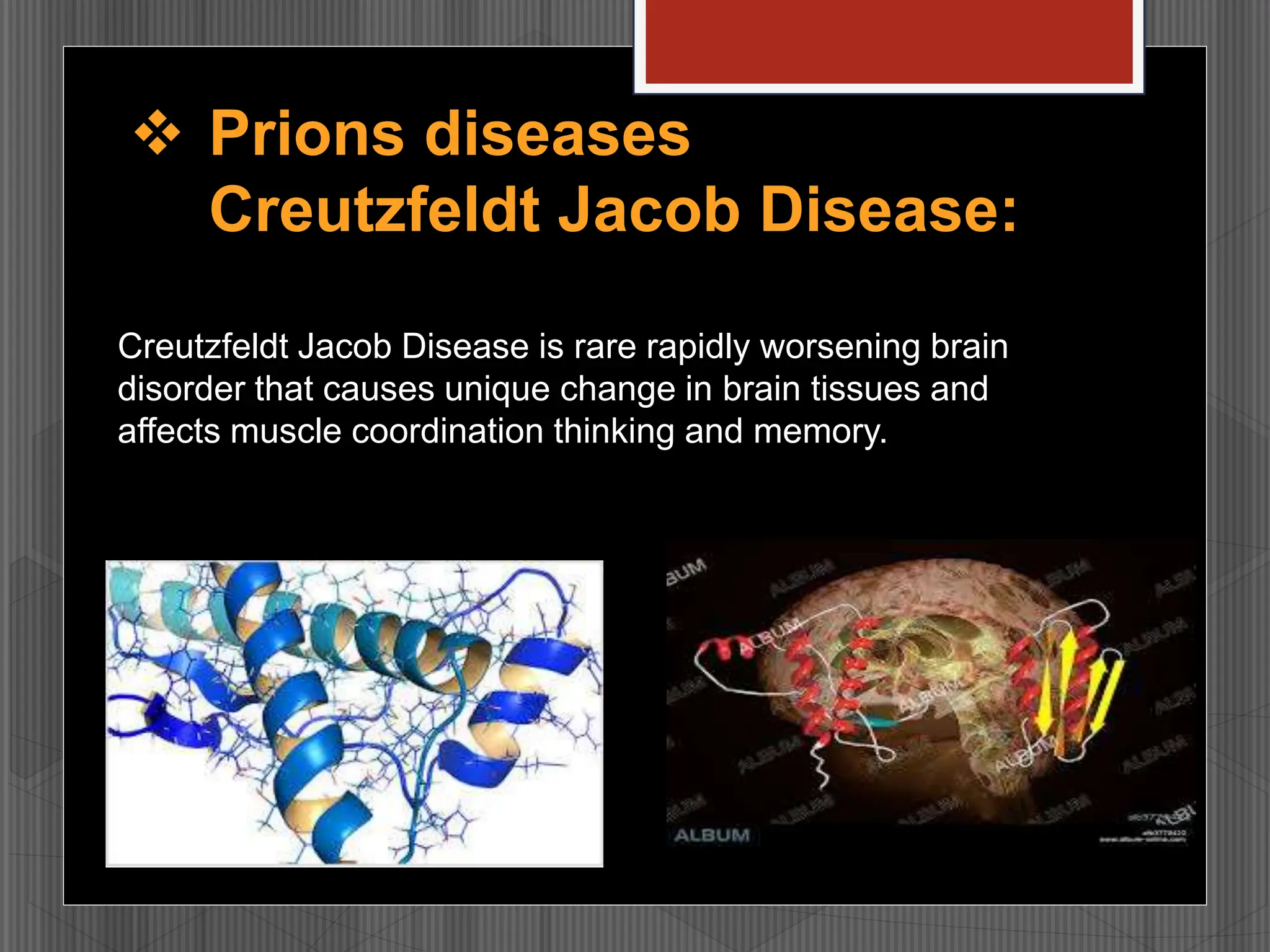
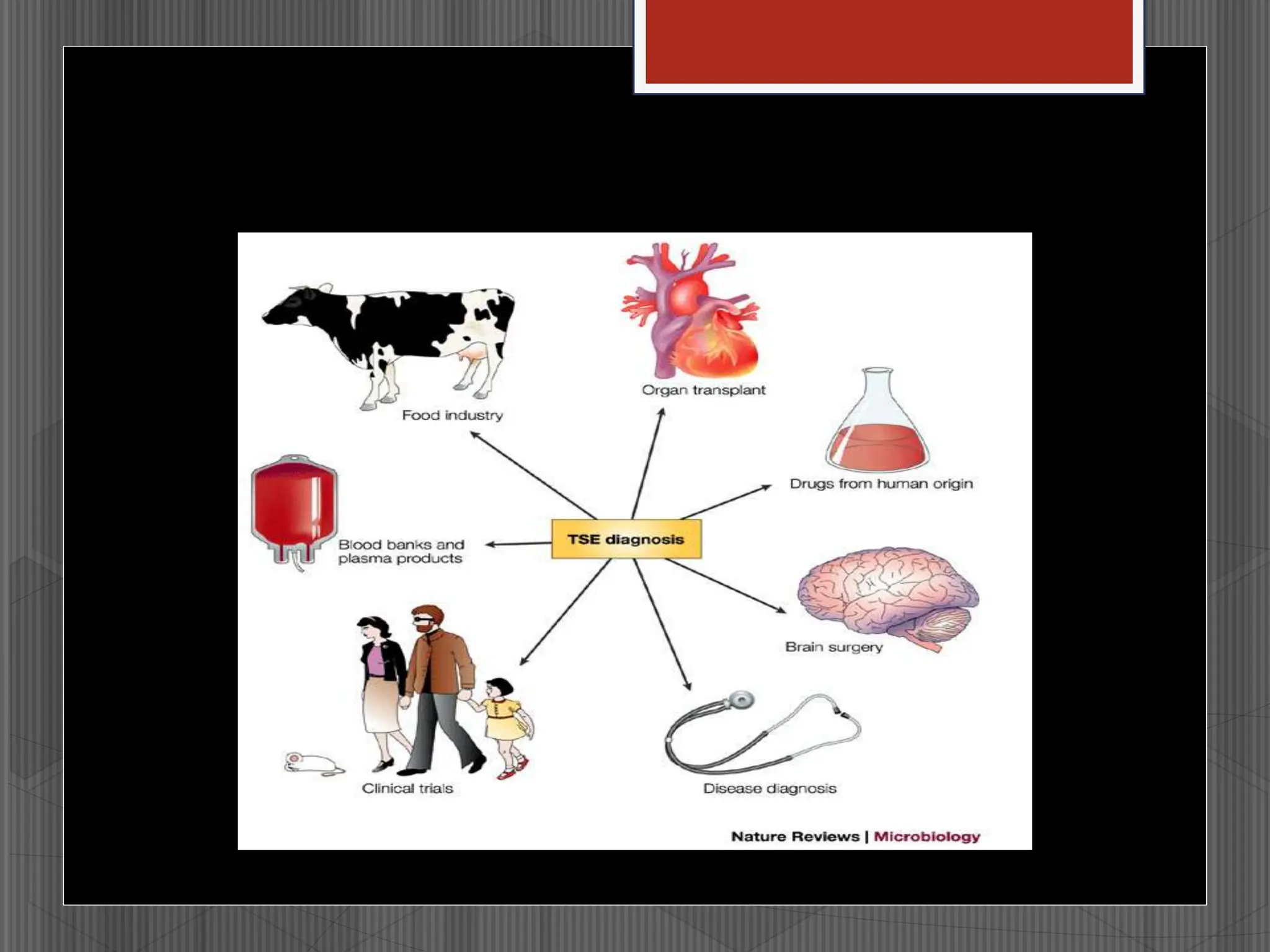
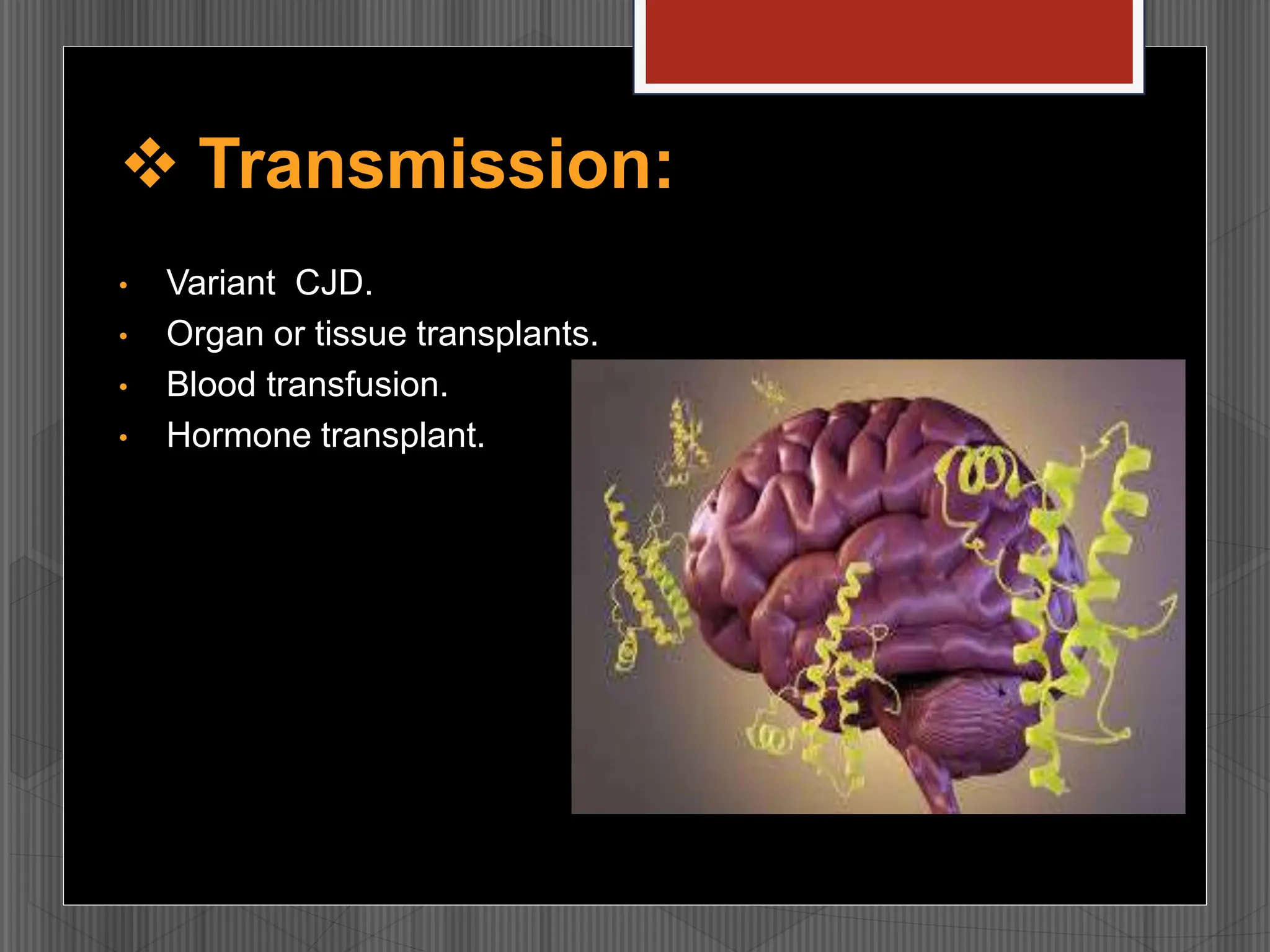
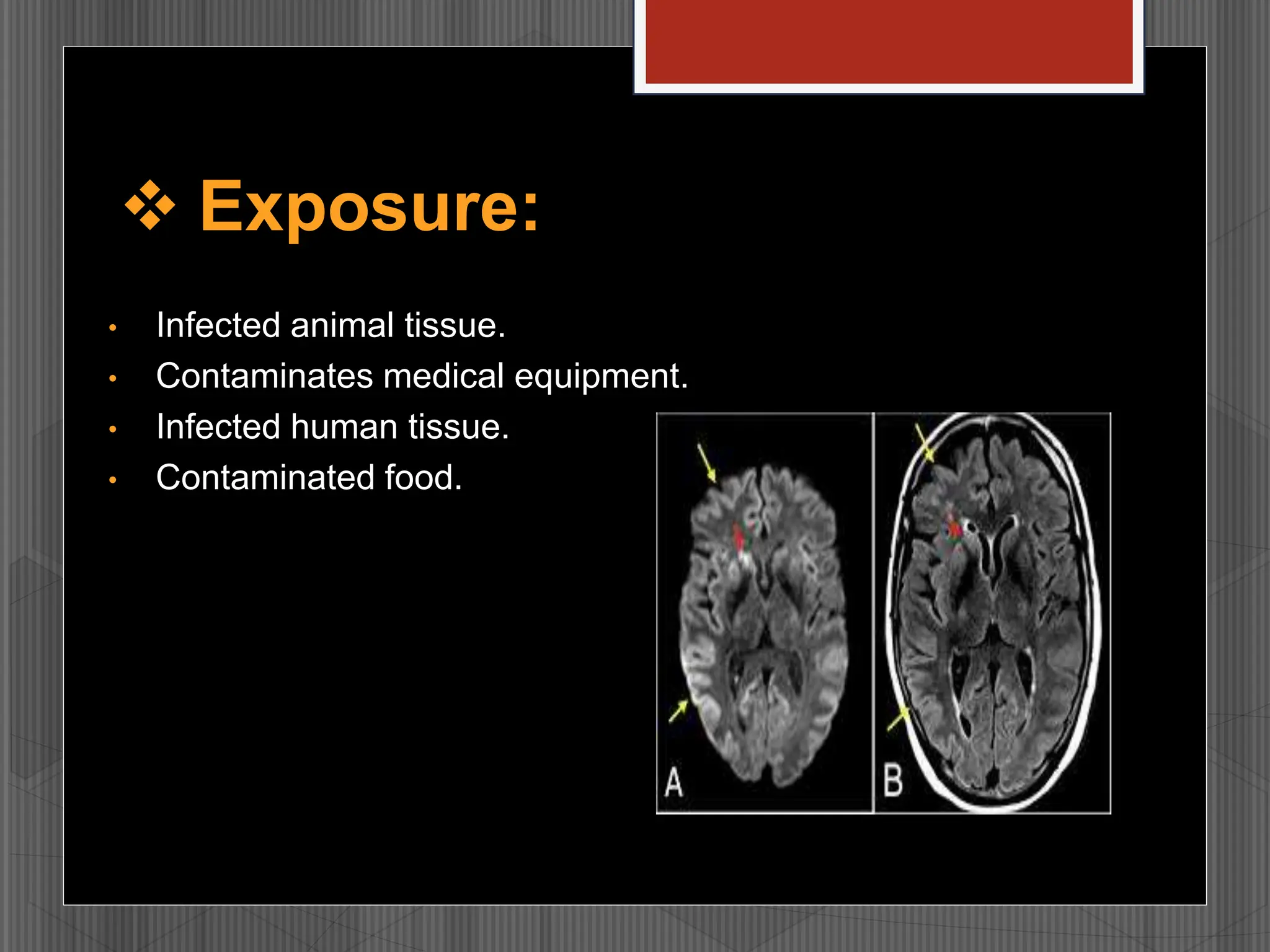
Prions are infectious proteins that cause fatal neurodegenerative diseases in humans and animals by inducing abnormal protein folding, leading to various disorders such as Creutzfeldt-Jakob disease and kuru. These diseases have distinct transmission routes, symptoms, and no known cures, with ongoing research aimed at understanding their mechanisms and developing effective treatments. The historical context includes notable discoveries and public health impacts, particularly related to mad cow disease and its transmission to humans.


































![When was the last recorded case of
• The end of cannibalism has put an end to kuru disease. T
case recorded was in 2005. [12] However, it remains the f
described transmissible spongiform encephalopathy unrav
mystery of other prion diseases.](https://image.slidesharecdn.com/prionsunfoldedproteins-240709154615-edd4954c/75/prions-types-disease-prevention-and-treatment-41-2048.jpg)



















































































