Downloaded 804 times







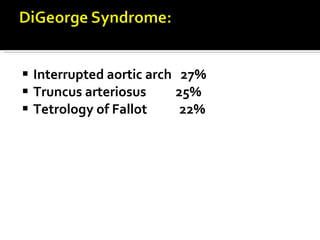







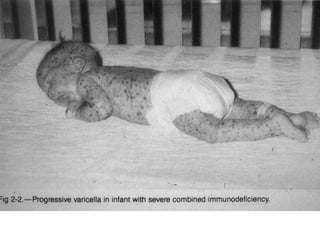













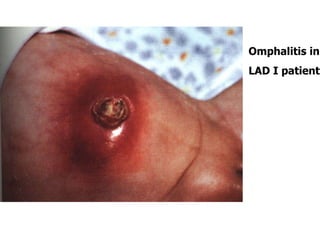








The document outlines various immunodeficiencies, including 'pure' T and B cell disorders, severe combined immunodeficiencies (SCID), and related genetic conditions. It describes the clinical manifestations, genetic factors, and potential treatments such as bone marrow transplantation and gene therapy. Specific syndromes like DiGeorge syndrome, X-linked SCID, and others are mentioned along with their respective symptoms and associated genetic anomalies.











































































































