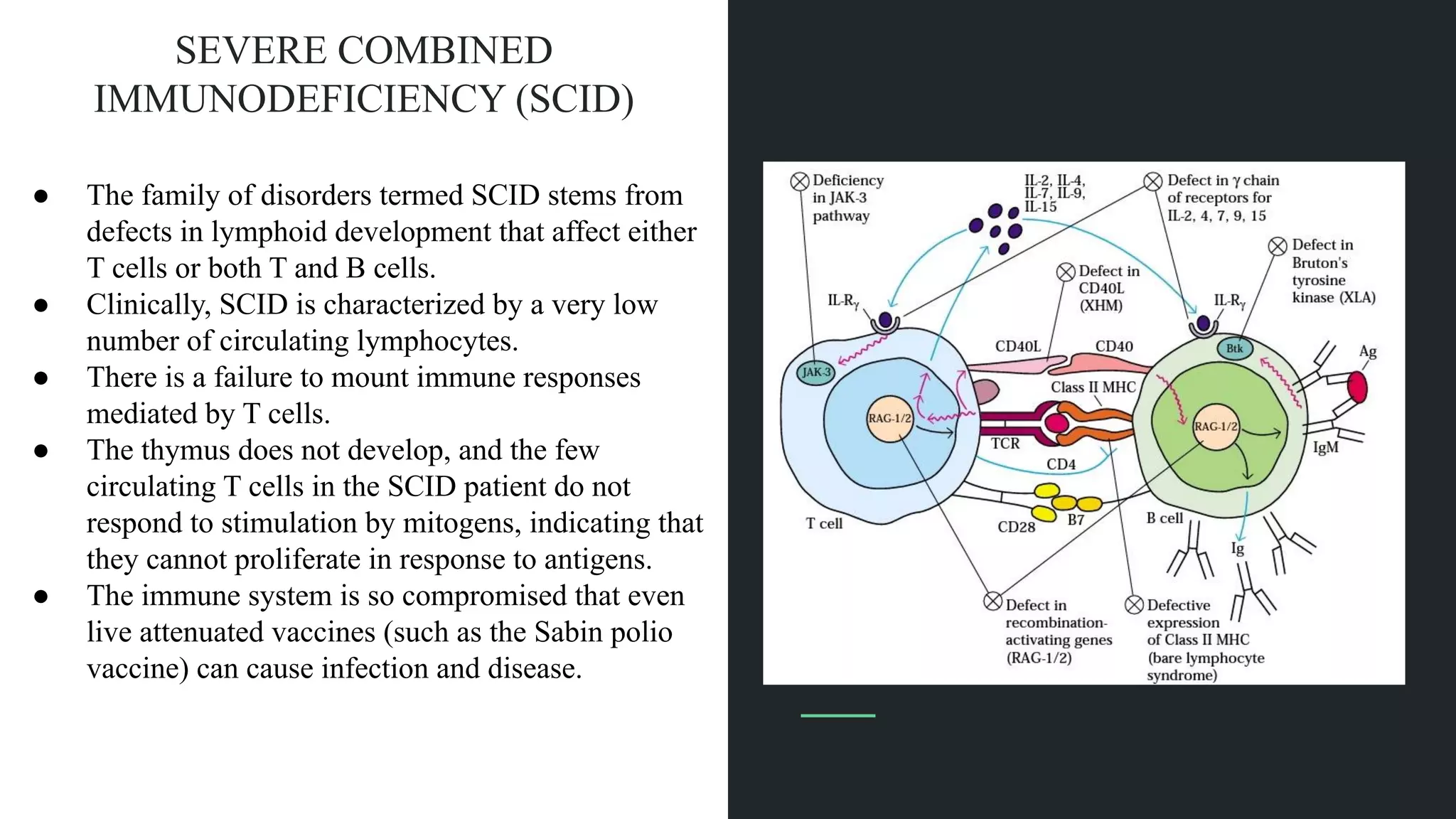
The document discusses various primary immunodeficiency diseases, focusing on their causes, symptoms, and genetic underpinnings. It covers conditions such as Severe Combined Immunodeficiency (SCID), Wiskott-Aldrich Syndrome, and Acquired Immunodeficiency Syndrome (AIDS), detailing how defects in the immune system can lead to increased susceptibility to infections and other health complications. Different immunodeficiencies are categorized based on their effects on adaptive or innate immunity and often involve genetic mutations that impact immune cell functionality.