Downloaded 1,524 times




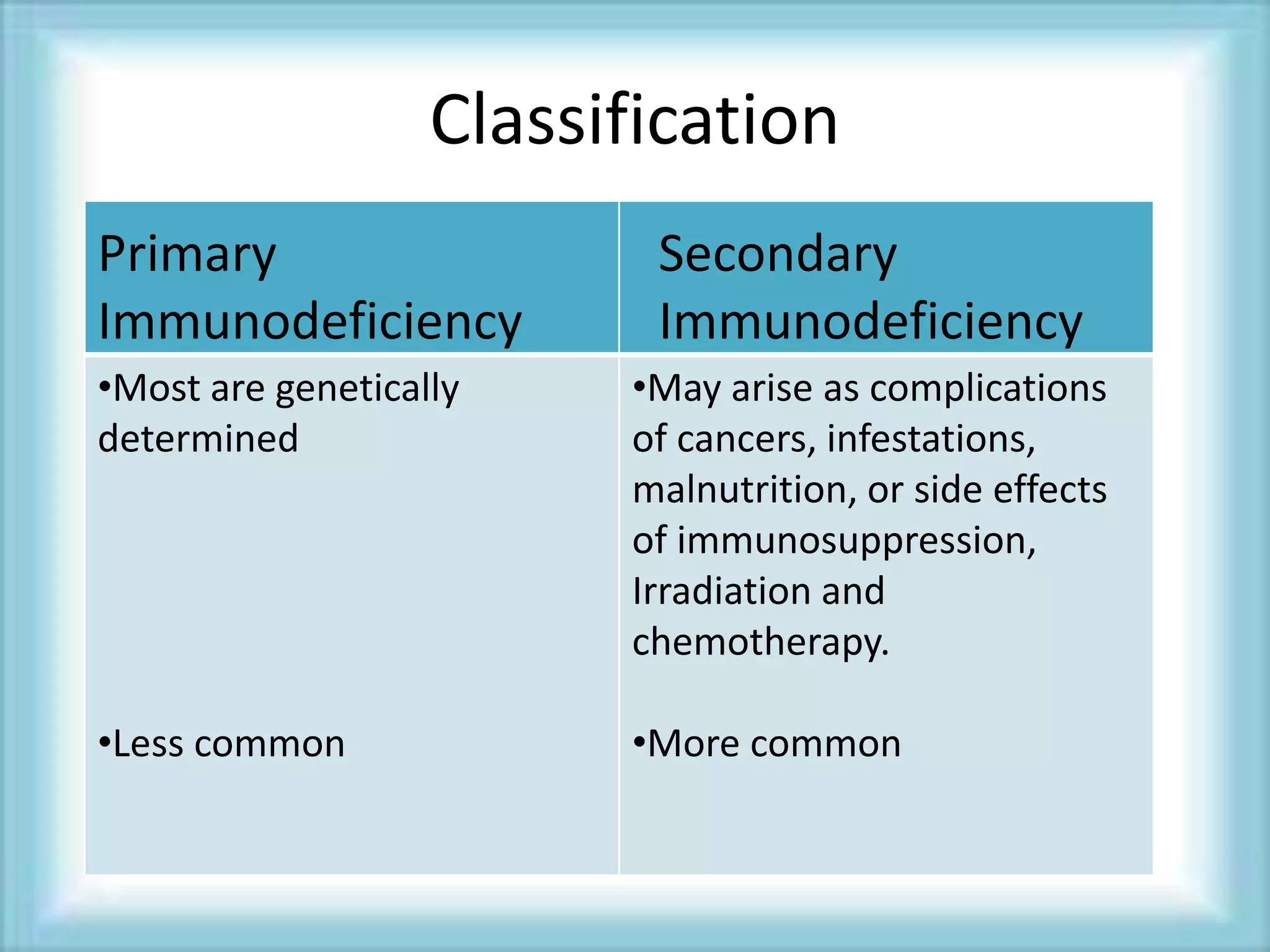


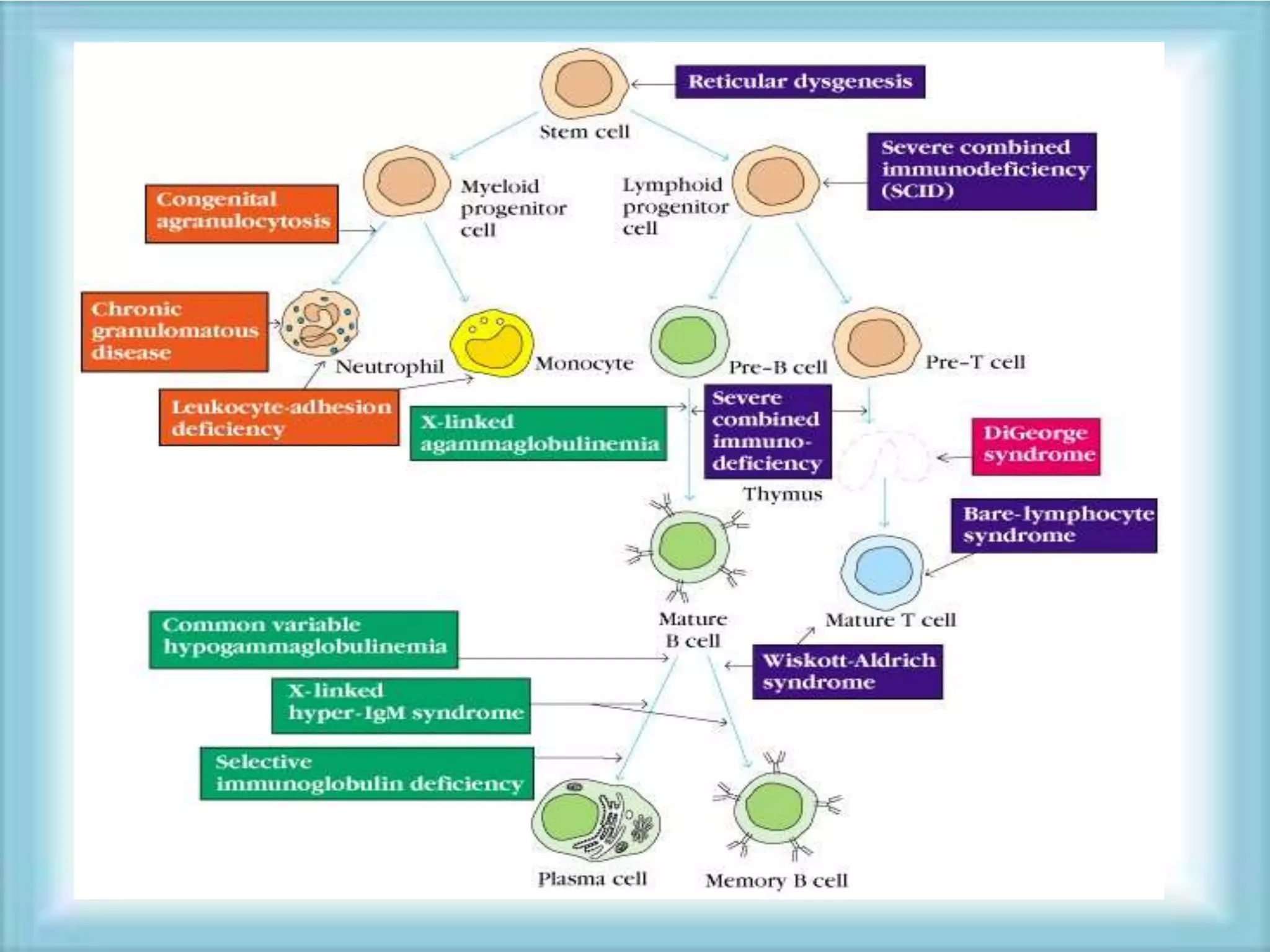




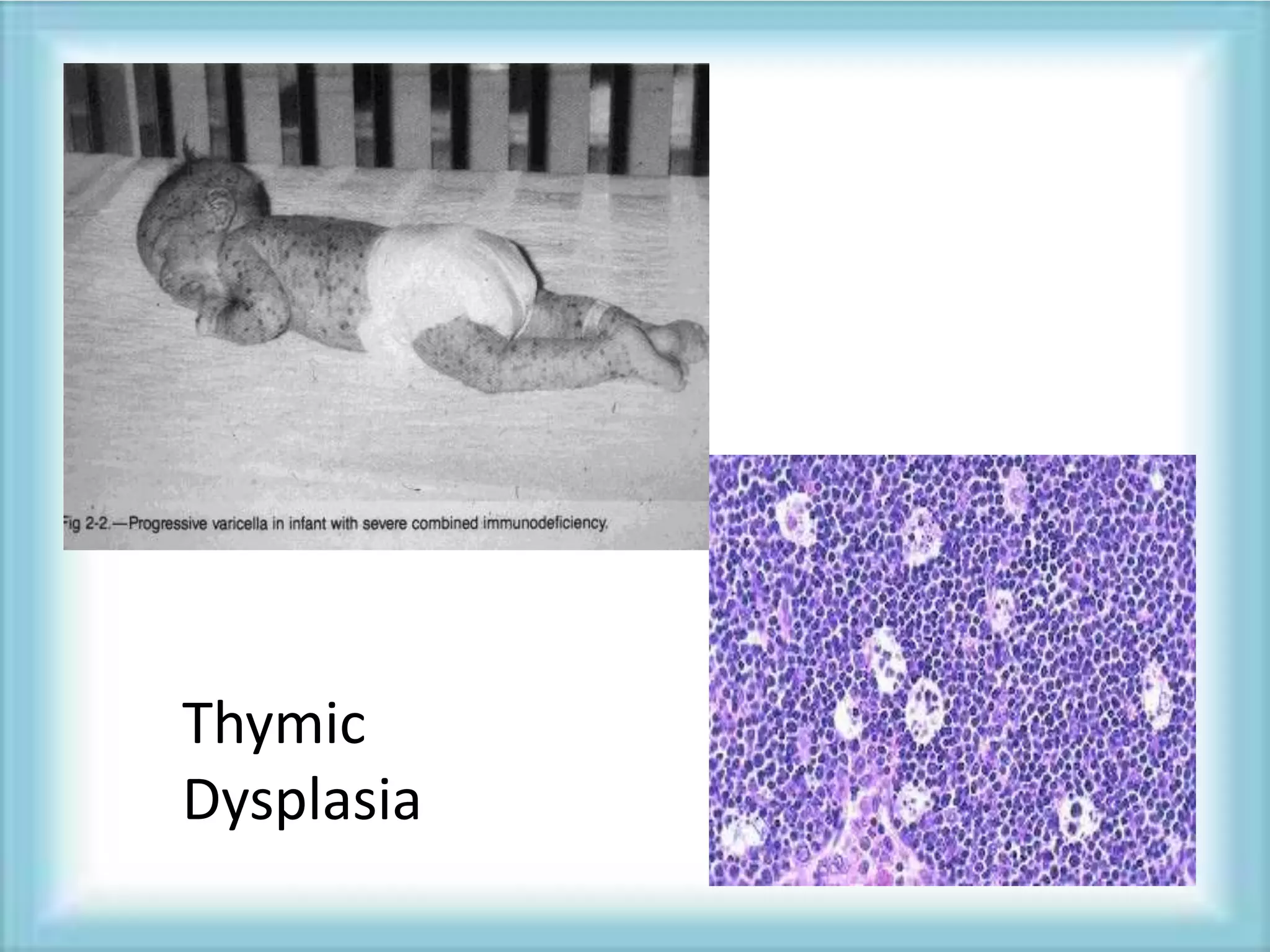


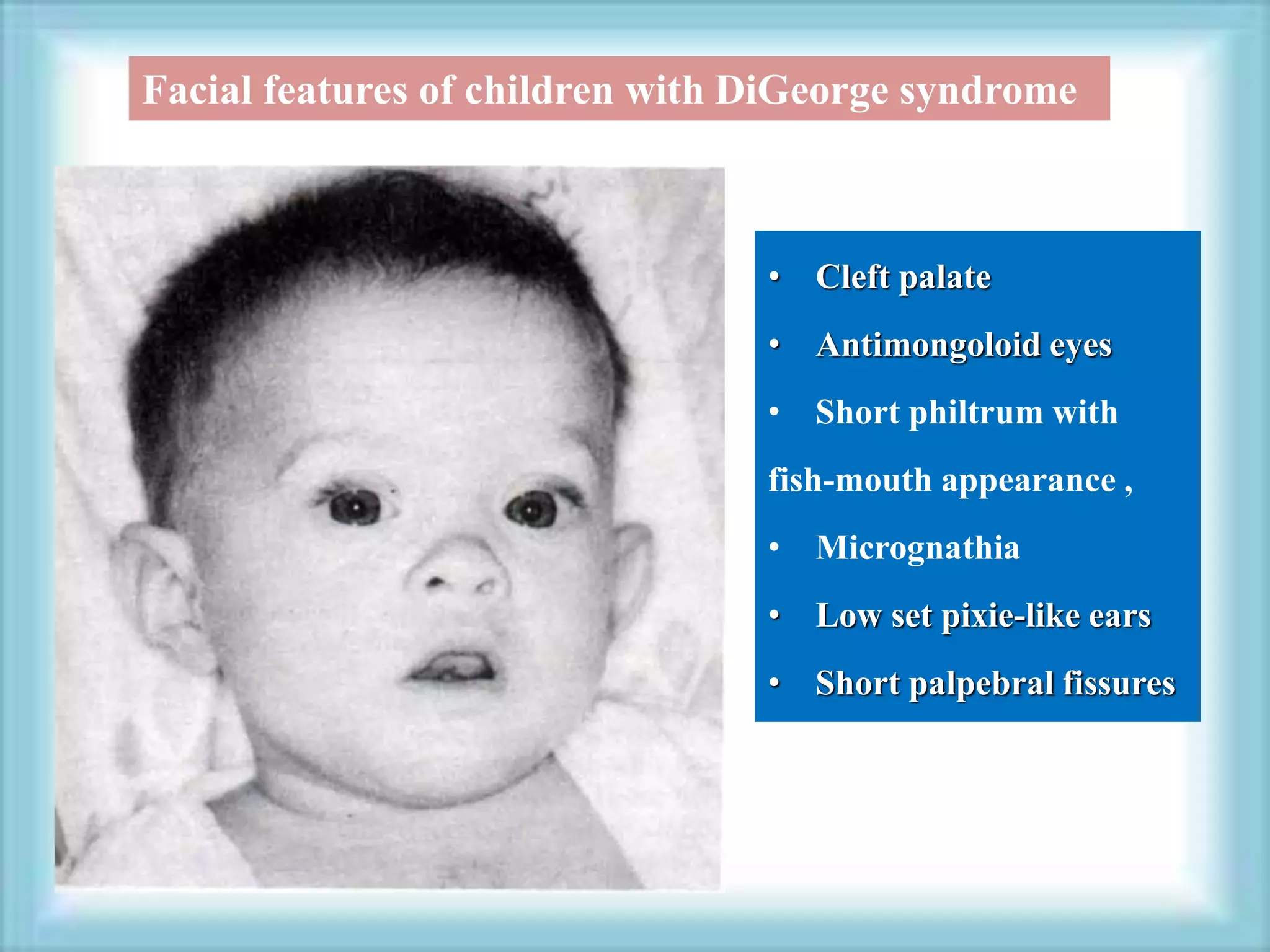
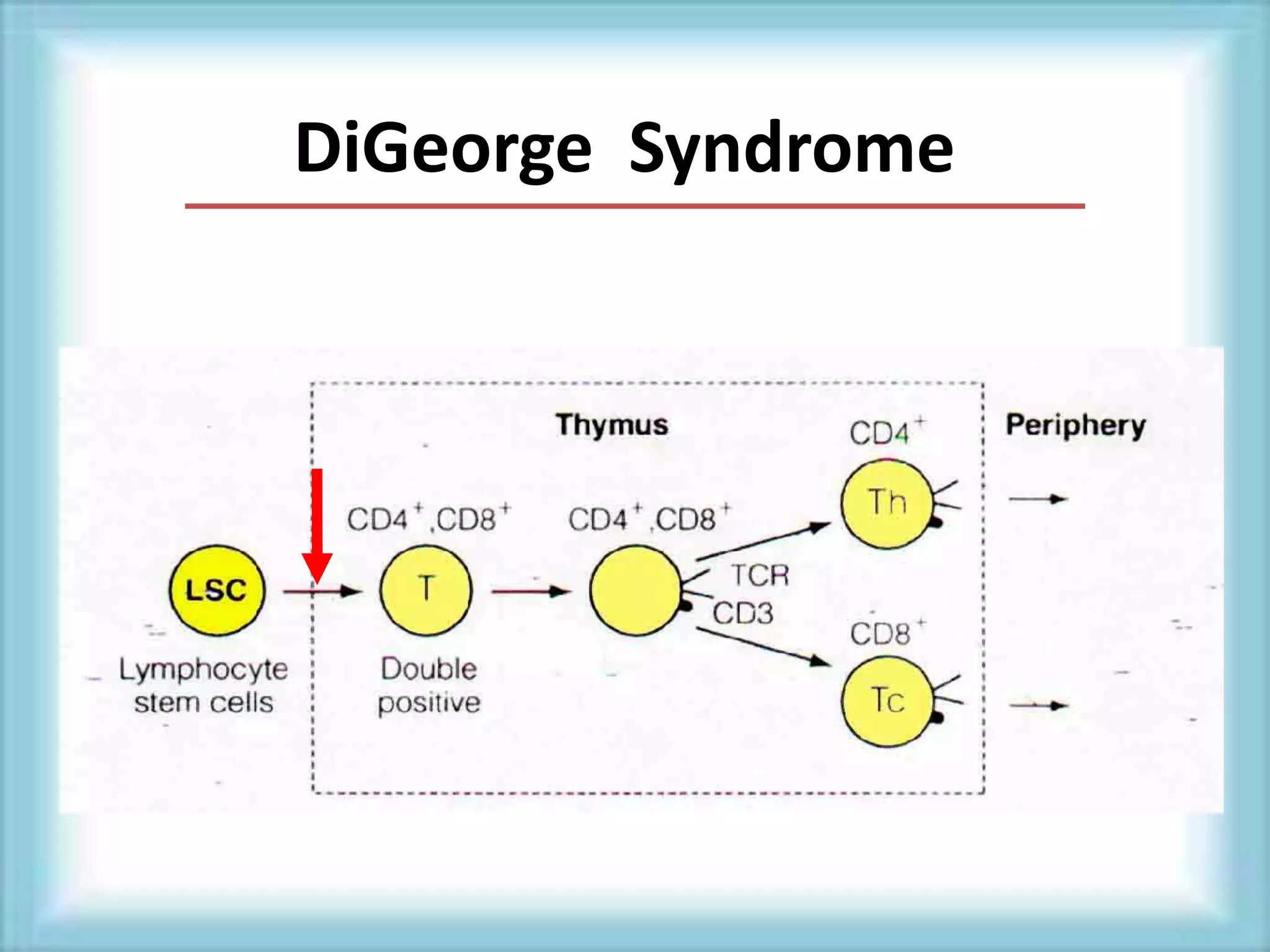
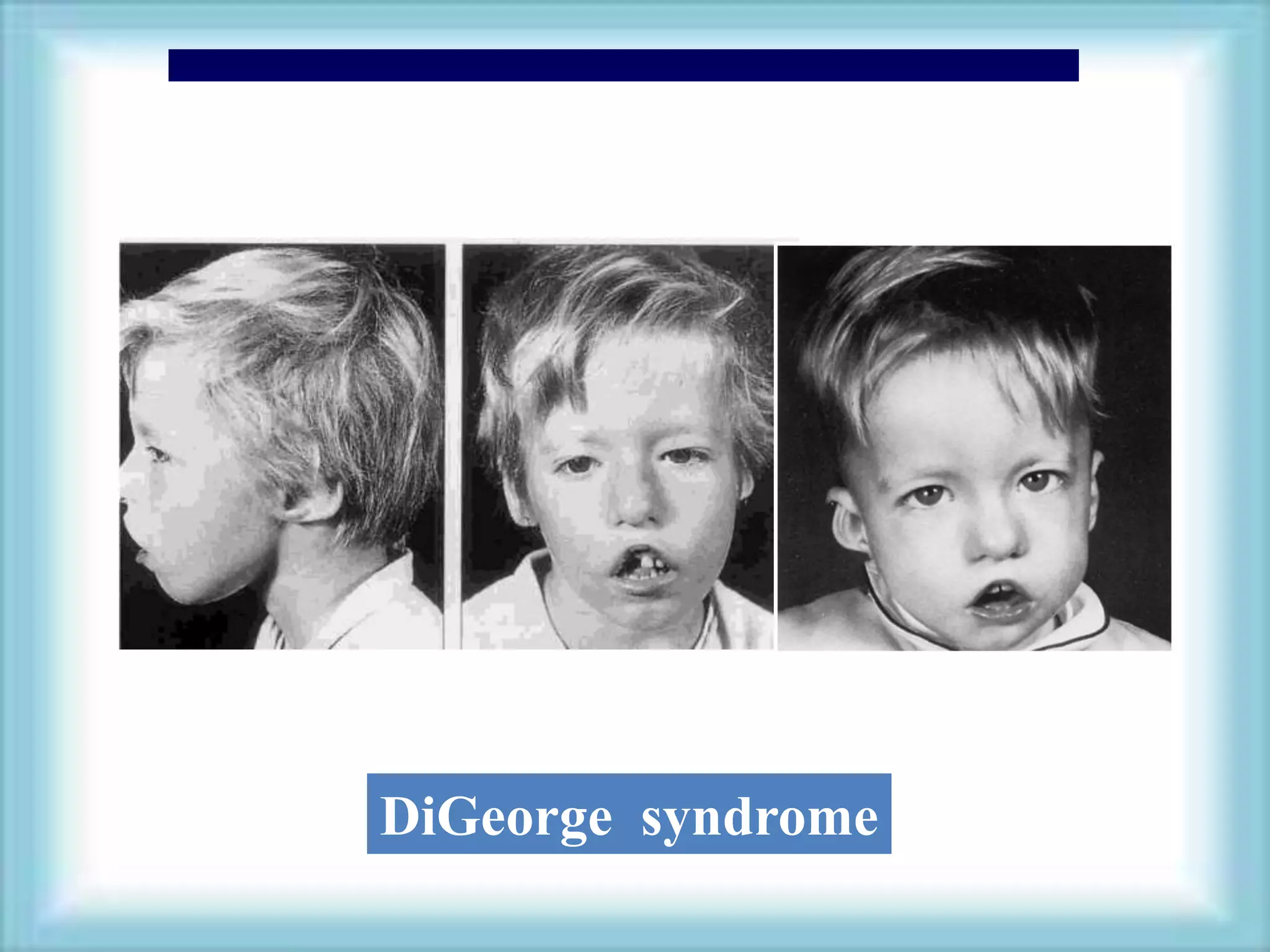

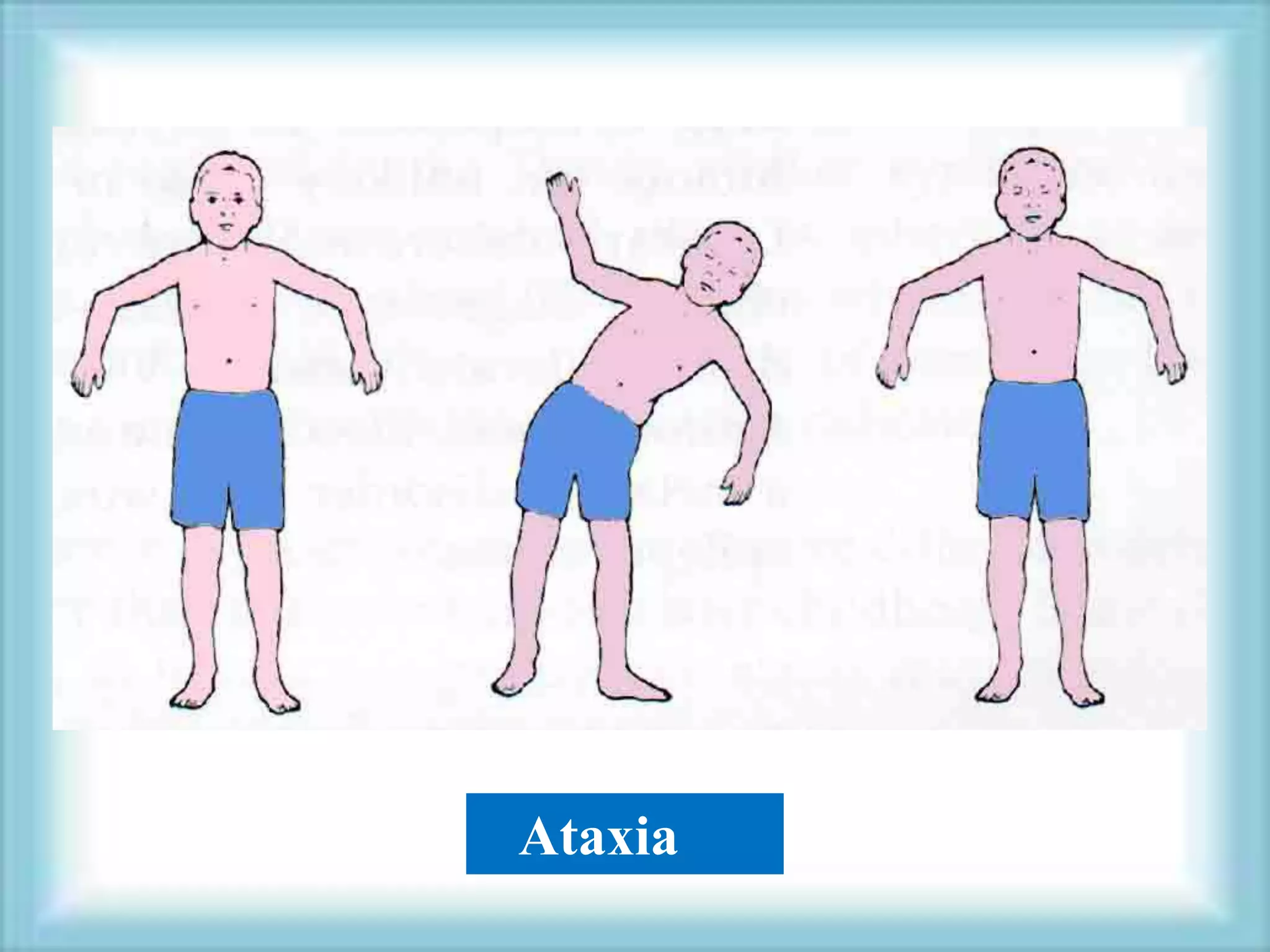
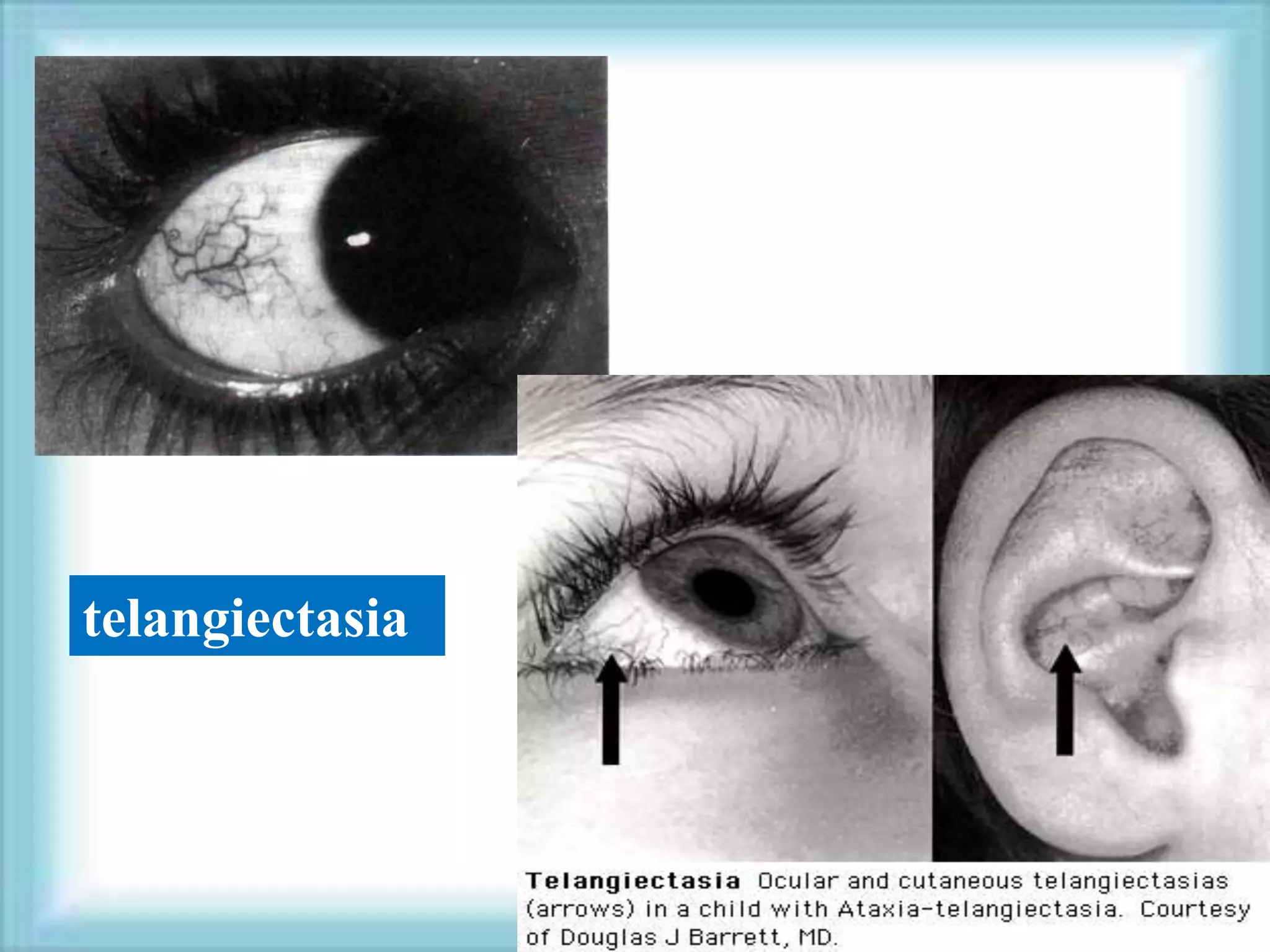

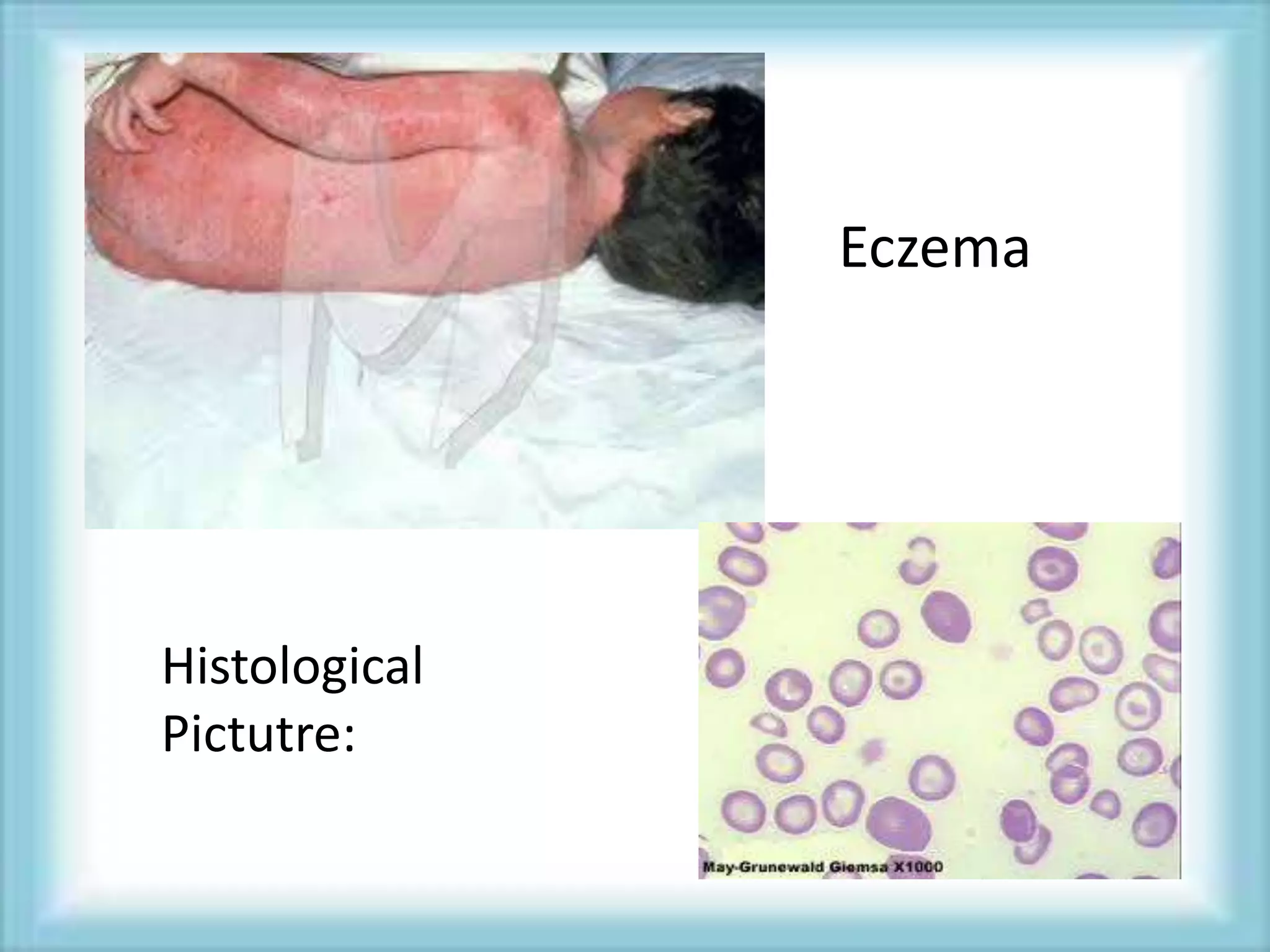

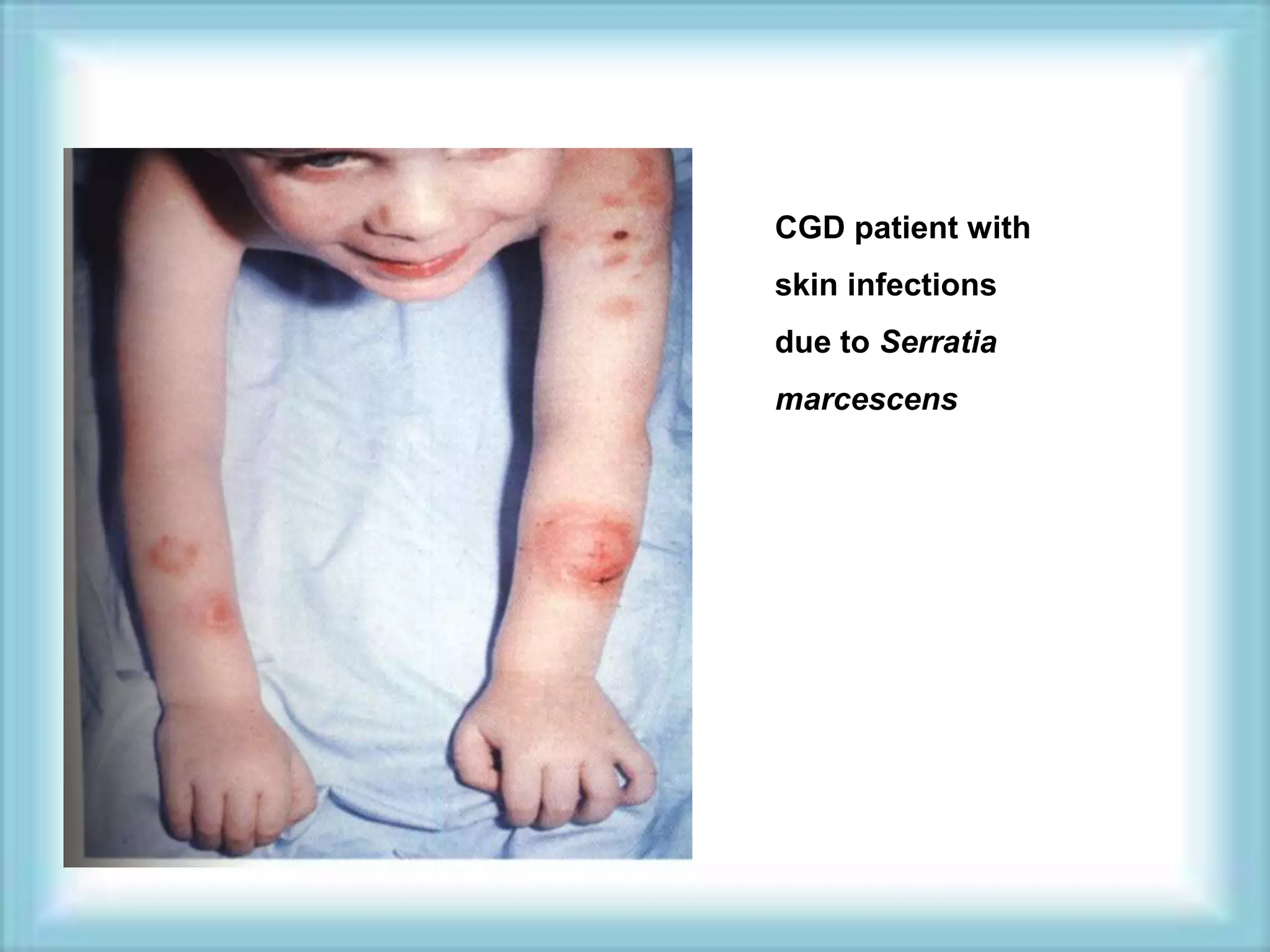



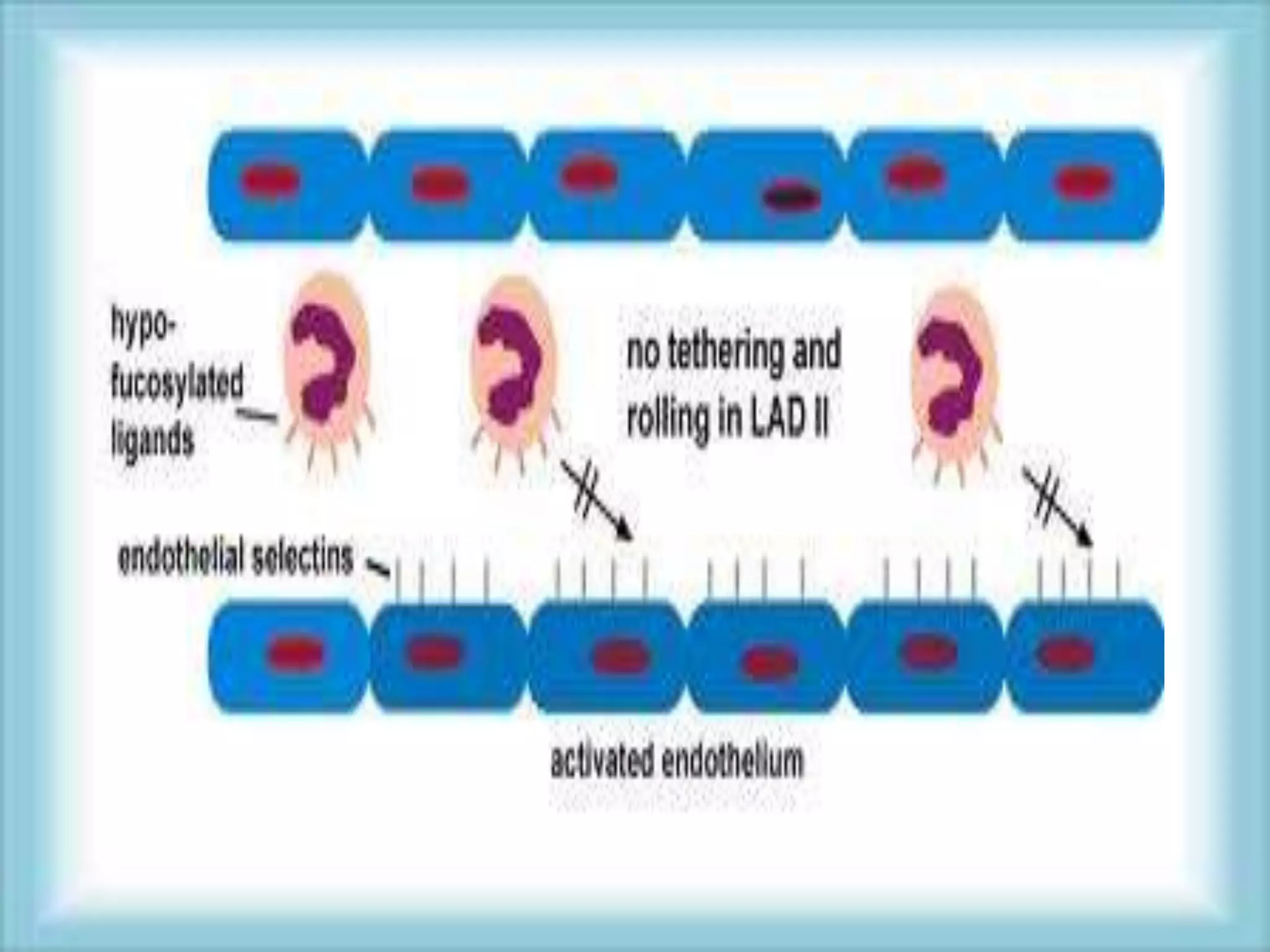






This document discusses primary immunodeficiencies, which are a group of genetically determined disorders characterized by impaired immune response. It defines several types of primary immunodeficiencies including SCID, XLA, DiGeorge syndrome, Ataxia-teleangectesia, Wiskott-Aldrich syndrome, and CGD. For each, it describes the genetic cause, characteristic infections, clinical features, and available therapies. The document provides an overview of primary immunodeficiencies for educational purposes.




























































































