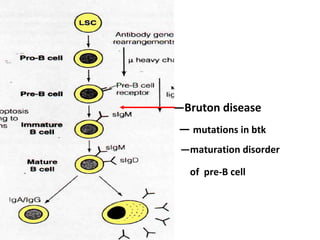
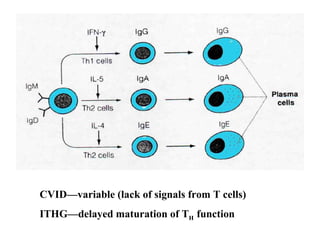
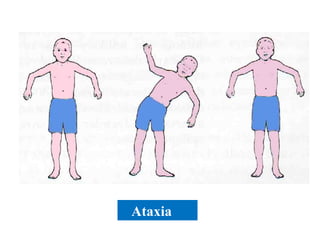
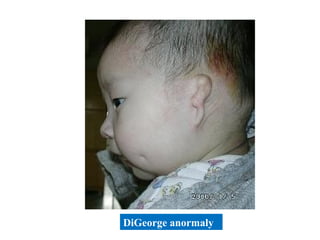
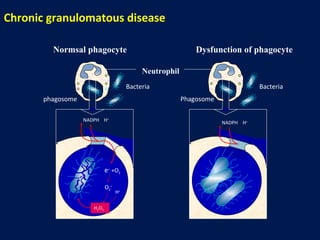
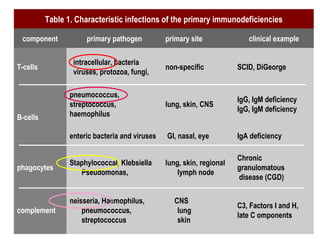
This document discusses a case presentation of a 2-year-old boy named D. George who has been brought in by his parents due to concerns about recurrent infections. The boy has a history of frequent upper respiratory infections and ear infections, and has been hospitalized twice for infections. The document provides background on primary immunodeficiency diseases and different aspects of the immune system to help evaluate the child's condition and determine if he has an immunodeficiency.