Downloaded 697 times



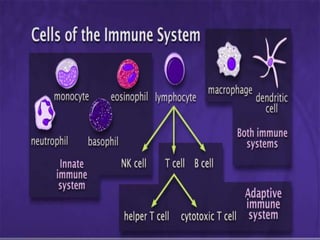







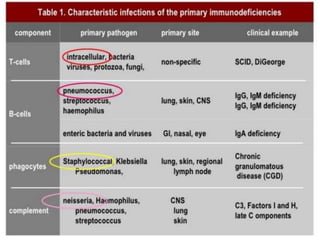

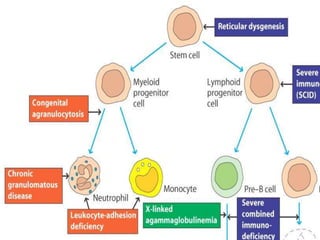
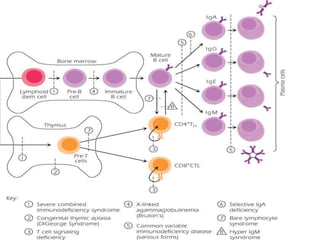



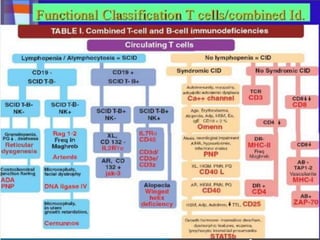
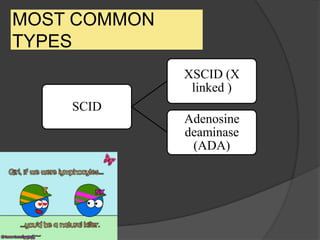



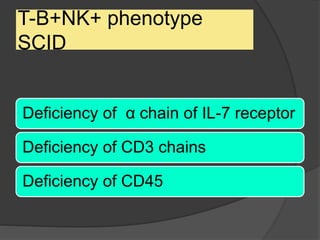





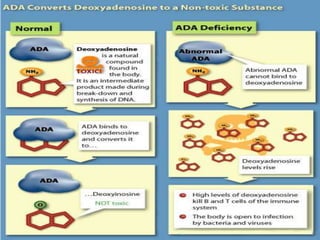
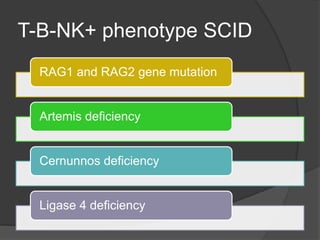



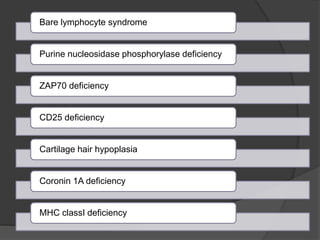




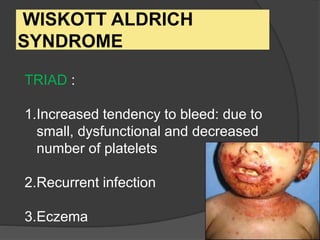


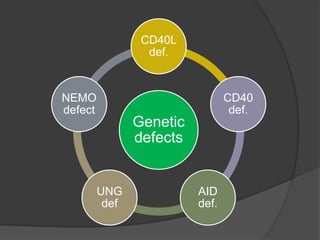
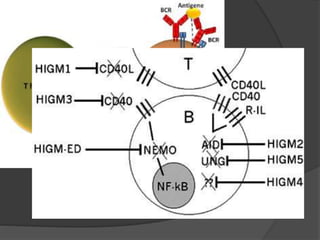



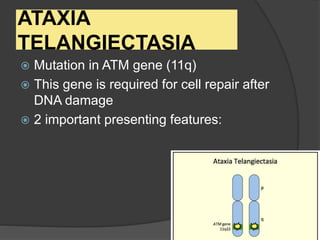
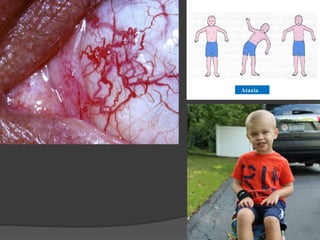









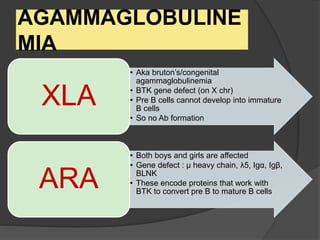
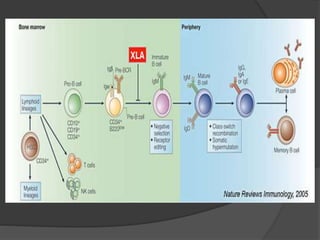
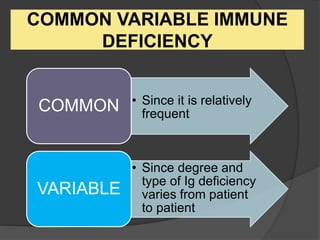
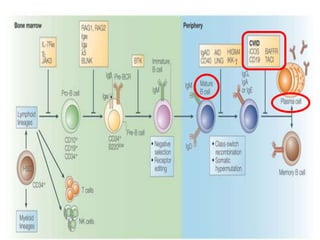
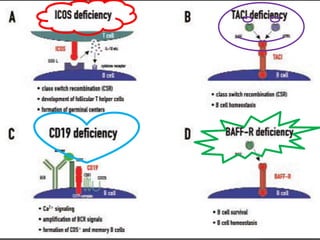

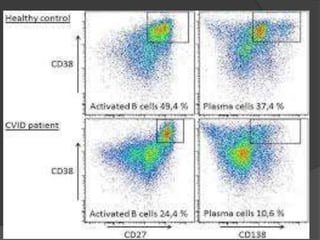

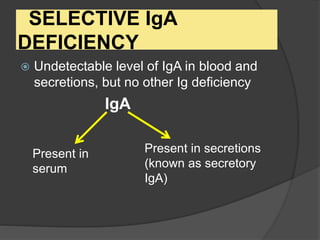













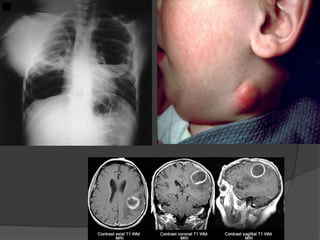

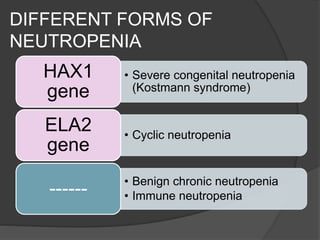
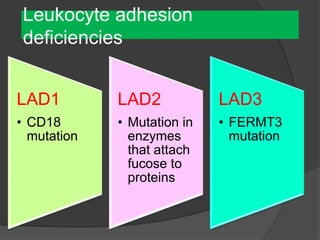
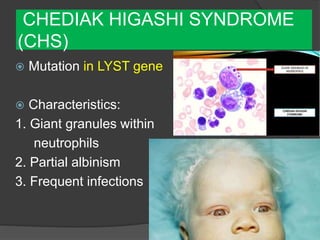

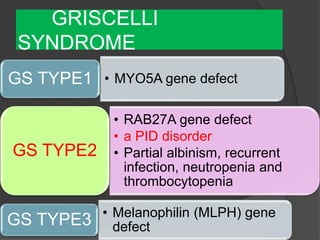


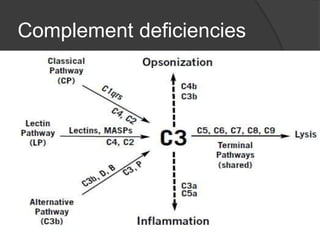







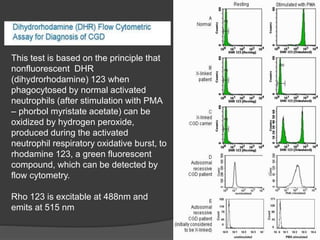







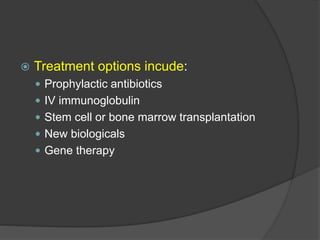




This document provides an overview of primary immunodeficiencies, including definitions, classifications, and details on specific deficiencies affecting B cells/antibodies, T cells, phagocytes, complement, and others. It discusses conditions like SCID, Wiskott-Aldrich syndrome, hyper-IgM syndrome, ataxia-telangiectasia, and DiGeorge syndrome. Diagnostic approaches like lymphocyte counts, genetic testing, and flow cytometry are also summarized.
















































































