Downloaded 11 times


































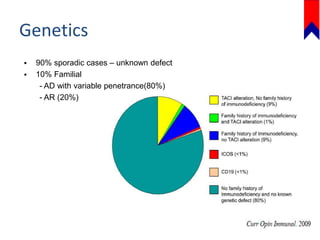

















This document discusses several primary immunodeficiencies including agammaglobulinemia, common variable immunodeficiency, and selective IgA deficiency. It provides information on the epidemiology, pathophysiology, clinical manifestations, diagnosis, and treatment of each condition. Key points include that agammaglobulinemia is caused by mutations in the BTK gene that halt B cell development, common variable immunodeficiency has unknown causes in most cases, and selective IgA deficiency involves a block in differentiation of B cells into IgA secreting plasma cells. Clinical features often involve recurrent respiratory and gastrointestinal infections. Treatment focuses on antibody replacement therapy and infection prophylaxis.






















































































