





![7
1.Overview:
Obesity is a serious medical condition whose prevalence is increasing in
developing countries also. This growing incidence represents a pandemic
that needs urgent attention if the potential morbidity, mortality, and
economic tolls that will be left in its wake are to be avoided. Obesity
predisposes to increased risk of a number of medical conditions including
type II diabetes mellitus, hypertension, coronary heart disease,
osteoarthritis, respiratory problems and cancers of breast, endometrium,
prostate, bowel cancers (1,2). Obesity represents a state of excess storage
of body fat. Although very similar, the term overweight is defined as an
excess body weight for height.
The body mass index (BMI), also known as the Quetelet index is a WHO
accepted index for classifying the degree of obesity. Standards defining
overweight and obesity on the basis of BMI were developed by the
International Obesity Task Force of the World Health Organization. BMI
= (weight [kg])/ (height[m] 2). Under this convention for adults, grade 1
overweight (commonly and simply called overweight) is a BMI of 25–
29.9 kg/m2. Grade 2 overweight (commonly called obesity) is a BMI of
30–39.9 kg/m2. Grade 3 overweight (commonly called severe or morbid
obesity) is a BMI greater than or equal to 40 kg/m2.
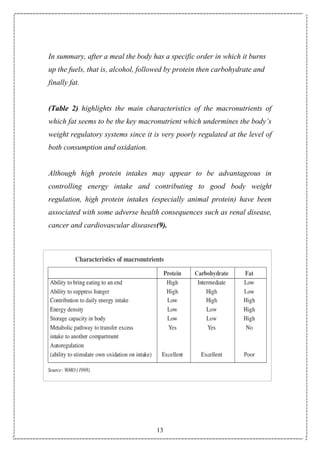
The laws of thermodynamics are applicable here also because if energy
expenditure by the body is less than the consumption, it will be stored in
the body in the form of adipose tissue. Appetite regulation is important
because it modulates the energy consumption side of the equation.
Appetite includes various aspects of eating patterns such as frequency
and size of eating episodes (gorging versus nibbling), choices of high fat](https://image.slidesharecdn.com/obesity1-150913223932-lva1-app6891/85/Obesity-7-320.jpg)








































![51
7.The Central Effects of Thyroid Hormones on Appetite:
7.1. Introduction:
Obesity, its complications, and the associated mortality are major public
health issues worldwide. The major central nervous system (CNS) areas
important in the regulation of appetite are the hypothalamus and
brainstem. The hypothalamus interprets and integrates afferent signals
from the periphery and that regulate food intake and energy expenditure.
brainstem to modulate efferent signals Neural and hormonal peripheral
signals communicate information including acute nutritional states and
energy stores. The hypothalamus is subdivided into a number of
interconnecting nuclei, including the paraventricular nucleus (PVN), the
ventromedial nucleus (VMN), and the arcuate nucleus(ARC), which are
particularly important in regulating energy homeostasis. The ARC is
located near the median eminence, where the blood-brain barrier is
incomplete, and is thus well positioned to respond to circulating factors
involved in appetite and food intake [170].
Recent evidence suggests that thyroid hormones may access the ARC and
other regions of the hypothalamus to regulate appetite (Figure 18). It is
well established that the hypothalamic-pituitary thyroid(HPT) axis
regulates body weight. Thyroid hormones are known to effect metabolic
rate. Thyroid dysfunction can have clinically significant consequences on
appetite and body weight. Hypothyroidism classically causes reduced
basal energy expenditure [171] with weight gain[172, 173]. Conversely,
hyperthyroidism increases energy expenditure and reduces body weight
[174–176]. Traditionally, it has been assumed that it is this reduced body
weight that drives the hyperphagia that can be a presenting feature in](https://image.slidesharecdn.com/obesity1-150913223932-lva1-app6891/85/Obesity-51-320.jpg)
![52
hyperthyroidism. However, recent evidence suggests that the HPT axis
may play a direct role in the hypothalamic regulation of appetite,
independent of effects on energy expenditure. Classically, hypothalamic
thyrotropin-releasing hormone (TRH) stimulates thyroid-stimulating
hormone TSH) release from the anterior pituitary gland, which then
stimulates the release of both thyroid hormones, triiodothyronine (T3)
and thyroxine (T4). Reports suggest that all of these signalling molecules
can directly influence food intake [177–180]. Improved understanding of
the role of the HPT axis and thyroid hormone in appetite may identify
new targets for anti obesity agents.
7.1.2. Effects of Thyroid Hormones on Food Intake:
There are well-characterised effects of fasting on hypothalamic TRH
expression. This is primarily thought to down regulate the HPT axis in
periods of limited food availability, thus reducing food intake. However,
TRH has been reported to have direct anorectic effects, suggesting it may
regulate food intake independent of effects on the HPT axis. In rodents,
central administration of TRH reduces food intake[177, 181, 182];
similar effects on food intake are seen following peripheral
administration [183].
TSH has also been shown to reduce food intake when injected centrally
into rats [177]. There is evidence that TSH from the pars tuberalis is
involved in the photoperiodic response in birds and rodents, and it is thus
possible that TSH is involved with the seasonal alterations in food intake
and body weight that occur in some species [184–186].](https://image.slidesharecdn.com/obesity1-150913223932-lva1-app6891/85/Obesity-52-320.jpg)
![53
The hyperphagia associated with hyperthyroidism may be a result of
thyroid hormones acting directly on CNS appetite circuits. T3 directly
stimulates food intake at the level of the hypothalamus. In rodent models,
peripheral and central hypothalamic administration of T3 increases food
intake [178–180].
There are several mechanisms postulated to mediate the orexigenic
effects of thyroid hormones. The ARC contains two distinct energy
homeostasis-regulating neuronal populations. One subpopulation
expresses the proopiomelanocortin(POMC) gene which codes for the
anorectic neuropeptide alpha-melanocyte-stimulating hormone(α-MSH).
The other expresses the orexigenic factors neuropeptide Y (NPY) and
agouti-related protein (AgRP). It has been reported that peripheral
administration of T3 increases hypothalamic NPY mRNA and that
intracerebroventricular(ICV) administration of a NPY Y1 receptor
antagonist blunts T3 induced hyperphagia, suggesting that T3 may
increase appetite via NPY [179]. T3 administration was also reported
toalso reduce hypothalamic POMC expression [179]. Another study did
not detect changes in hypothalamic neuropeptide expression in response
to peripheral administration of T3 though thismay reflect the different
doses of T3 administered[178].](https://image.slidesharecdn.com/obesity1-150913223932-lva1-app6891/85/Obesity-53-320.jpg)
![54
(table 4)
However, the effects of thyroid hormones on food intake may not be
mediated directly by the ARC. Direct administration of T3 into the VMN
but not the ARC increases food intake in rats [178]. As appetite
regulating circuits in the ARC are known to be altered by changes in the
HPT, there may be an indirect effect of the ARC via the VMN allowing
intra-VMN T3 to increase food intake. In keeping with this, there are
excitatory inputs into POMC neurons that originate in the VMN [187].](https://image.slidesharecdn.com/obesity1-150913223932-lva1-app6891/85/Obesity-54-320.jpg)
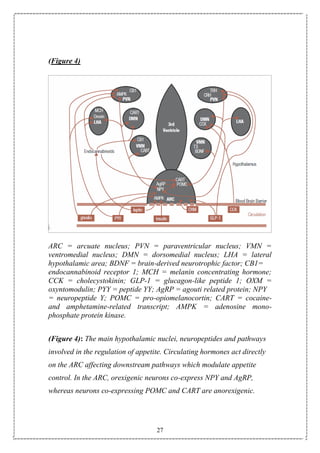
![55
(Figure 18)
(Figure 18): Schematic diagram of central appetite regulation. T3 can
access the hypothalamus and brainstem via the incomplete blood brain
barrier. PVN: paraventricular nucleus; ARC: arcuate nucleus; VMN:
ventromedial nucleus; BBB: blood-brain barrier; T3: triiodothyronine;
POMC: Pro-opiomelanocortin; NPY: neuropeptide Y; AgRP: agouti-
related protein; BDNF: brain-derived neurotrophic factor; HPT:
hypothalamic-pituitary thyroid; SNS: sympathetic nervous system.
7.1.3 Effects of Nutritional State on Thyroid Hormones:
Reduction in TRH in response to fasting may be important as TRH is seen
to have a direct anorectic effect when injected into the hypothalamus
[182]. It is possible there are distinct TRH neuronal populations
regulating the HPT axis and regulating appetite.
In periods of limited food availability, there is central downregulation of
the HPT axis. Serum T4 and T3 levels fall during fasting in humans [188]](https://image.slidesharecdn.com/obesity1-150913223932-lva1-app6891/85/Obesity-55-320.jpg)
![56
and rodents [189, 190]. As the majority of T3 in rodents comes from the
thyroid gland, it is thought food deprivation may result in a fall in the
release of T4 and T3. This is likely secondary to a reduction in
hypothalamic TRH expression, an effect that may be mediated by the
adipose hormone lepton.
(Figure19)
(Figure19): Effect of fasting on the hypothalamo-pituitary-thyroid axis.
PVN: paraventricular nucleus; ARC: arcuate nucleus; TRH: thyrotropin
releasing hormone; TSH: Thyroid-stimulating hormone; T3:
triiodothyronine; T4: thyroxine; POMC: Pro-opiomelanocortin; NPY:
neuropeptide Y; AgRP: agouti-related protein.](https://image.slidesharecdn.com/obesity1-150913223932-lva1-app6891/85/Obesity-56-320.jpg)
![57
8.Growth Hormone (GH) secretion:
8.1. Introduction
Human growth hormone (GH) is a mixture of peptides, the major
physiologic and bio active component being a 22 kDa polypeptide chain
of 191 amino acids secreted by the anterior pituitary gland . In man GH is
secreted episodically in a pulsatile fashion. The main regulatory
hormones of GH are two hypothalamic peptide hormones: GH releasing
hormone (GHRH) a 44 amino-acid peptide required for the initiation of
GH pulses and somatostatin an inhibitory peptide which modulates the
amplitude of GH pulses. However, several brain transmitter pathways as
well as sleep and several other factors seem to be involved in GH
regulation, suppressing or stimulating GH release by influencing GHRH
or somatostatin .
8.2.Metabolic and nutritional factors:
8.2.1Glucose:
An impaired GH response to hypoglycaemia is well documented in
obesity.[191-193] Moreover, recent studies performed with
hyperglycaemic clamp and oral glucose load have demonstrated that in
obese patients, contrary to normal subjects, hyperglycaemia does not
inhibit spontaneous[194] and stimulated (GHRH, arginine,
hexarelin)[195,196] GH secretion. On the contrary, the somatotropin
response to GHRH and arginine isphysiologically blunted by](https://image.slidesharecdn.com/obesity1-150913223932-lva1-app6891/85/Obesity-57-320.jpg)
![58
administration of SRIH and of the cholinergic antagonist
pirenzepine.[196] These observations suggest an inability of hyper-
glycaemia to trigger hypothalamic SRIH release in obesity.
8.2.2.Insulin:
Obesity is characterized by fasting hyperinsulinemia and exaggerated
insulin release in response to a mixed and exaggerated insulin release in
response to a mixed meal or a glucose load.[197,198] and exaggerated
insulin release in response to a mixed meal or a glucose load.[197,198]
Experimental data support the existence of a negative feedback exerted
by circulating insulin on GH secretion. In normal subjects, a progressive
reduction of the GH response to hypoglycaemia[199] and GHRH[200]
has been observed with increasing insulin concentrations. The
mechanisms whereby insulin regulates GH release are not completely
clarified yet.
Insulin might act at both the hypothalamic and the pituitary level via its
multiple metabolic pathways. By binding to specific hypothalamic
receptors,[201 – 203] insulin could enhance the release of
catecholamines,[204,205] which in turn might stimulate SRIH discharge
via b-adrenergic receptors.[206]. However, in spite of the low number of
specific insulin receptors in normal pituitary cells,[207]an inhibition of
GH synthesis and release, along with a reduction of GH mRNA content in
somatotropes have been observed following exposure of these cells to
insulin in vitro.(208) Insulin might also regulate GH secretion through its
effects on aminoacid metabolism and ion transport. Lastly, insulin may
indirectly influence GH secretion by inhibition of IGFBP-1[209] and
hence by increasing the levels of free plasma IGF-I which negatively
feeds back on GH secretion. In spite of the above, the pathophysiological](https://image.slidesharecdn.com/obesity1-150913223932-lva1-app6891/85/Obesity-58-320.jpg)
![59
relevance of hyperinsulinaemia in the GH hyposecretion of obesity is
challenged by the observation that GH secretion is normal in diseases
other than obesity associated with high insulin levels[210] and that in
obese subjects, normalization of serum insulin is not followed by
restoration of normal GH secretion.
8.2.3.Aminoacids:
GH is known to stimulate aminoacid uptake and protein synthesis.[211]
In turn, aminoacids participate in the regulation of GH release. Indeed,
high protein meals and administration of basic (arginine and ornithine)
and aromatic (tryptophan) aminoacids, stimulate GH secretion in normal
subjects,[212,213] probably because of a decrease in hypothalamic
SRIH.114 As mentioned above, an impairment of the GH response to
arginine, alone or combined with GHRH, is well documented in obesity.](https://image.slidesharecdn.com/obesity1-150913223932-lva1-app6891/85/Obesity-59-320.jpg)













![73
121. Cvetkovic, V.; Brischoux, F.; Griffond, B.; Bernard, G.; Jacquemard, C.;Fellmann, D.
and Risold, P.Y.(2003): Evidence of melanin concentrating hormone-containing neurons
supplying both cortical and neuroendocrine projections. Neuroscience 116 (1), 31–35.
122. Ludwig, D.S., Tritos, N.A., Mastaitis, J.W., Kulkarni, R., Kokkotou, E.G., Elmquist,
J.L., Bradford, F., Jeffrey, S. and Maratos-Flier, E. (2001): Melanin-concentrating hormone
over-expression in transgenic mice leads to obesity and insulin resistance. J. Clin. Invest. 107,
379–386.
123. Katsuura, G. and Inui, A. (2003): Melanin-concentrating hormone as a metabolic and
cognitive regulatory factor. Curr. Med. Chem. – Central Nervous System Agents 3 (3), 217–
227.
124. Xu, R., Li, S., Paruchova, J., McBriar, M.D., Guzik, H., Palani, A., Clader, J.W., Cox,
K., Greenlee, W.J., Hawes, B.E., Kowalski, T.J., O’Neill, K., Spar, B.D., Weig, B. and
Weston, D.J.(2006): Bicyclic[4.1.0]heptanes as phenyl replacements for melanin
concentrating hormone receptor antagonists. Bioorg. Med. Chem. 14(10), 3285–3299.
125. Kowalski, T.J.; Spar, B.D.; Weig, B.; Farley, C.; Cook, J.; Ghibaudi, L.; Fried, S.,
O’Neill, K.; Del Vecchio, R.A.; McBriar, M.; Guzik, H.; Clader, J.; Hawes, B.E. and Hwa,
J.( 2006): Effects of a selective melanin-concentrating hormone 1 receptor antagonist on food
intake and energy homeostasis in diet-induced obese mice. Eur. J. Pharmacol. 535(1–3), 182-
191.
126. McBriar, M.D., Guzik, H., Shapiro, S., Paruchova, J., Xu, R., Palani, A., Clader, J.W.,
Cox, K., Greenlee, W.J., Hawes, B.E., Kowalski, T.J., O’neill, K., Spar, B.D., Weig, B.,
Weston, D.J., Farley, C. and Cook, J.(2006): Discovery of orally efficacious melanin-
concentrating hormone receptor-1 antagonists as antiobesity agents. Synthesis, SAR, and
biological evaluation of bicyclo[3.1.0]hexyl ureas. J. Med. Chem. 49 (7), 2294–2310.
127. Williams, G., Joanne, A., Harrold and Cutler, D.J. (2000): The hypothalamus and the
regulation of energy homeostasis: Lifting the lid on the black box. Proc. Nutr. Soc. 59, 385–
396.
128. Allen, Y.S.; Adrian, T.E.; Allen, J.M.; Tatemoto, K.; Crow, T.J.; Bloom, S.R. and Polak
, J.M. (1983): Neuropeptide Y distribution in the rat brain. Science 221, 877–879.
129. Edwards, C.M.; Abusnana, S.; Sunter, D.; Murphy, K.G.; Ghatei, M.A. and Bloom, S.R.
(1999): The effect of the orexins on food intake: comparison with neuropeptide Y, melanin-
concentrating hormone and galanin. J. Endocrinol. 3, R7–R12.
130. Kalra, S.P. and Kalra, P.S. (2004): NPY and cohorts in regulating appetite, obesity and
metabolic syndrome: beneficial effects of gene therapy. Neuropeptides 38 (4), 201–211.
131. Pedrazzini, T. (2004): Importance of NPY Y1 receptor-mediated pathways: assessment
using NPY Y1 receptor knockouts. Neuropeptides 38 (4), 267–275.
132. Inui, A. (2000): Transgenic approach to the study of body weight regulation. Pharmacol.
Rev. 52 (1), 35–62.
133. Clark, J.T.; Kalra, P.S.; Crowley, W.R. and Kalra, S.P.( 1984): Neuropeptide Y and
human pancreatic polypeptide stimulate feeding behavior in rats. Endocrinology 115, 427–
429.](https://image.slidesharecdn.com/obesity1-150913223932-lva1-app6891/85/Obesity-73-320.jpg)








This document discusses the relationship between hormones and obesity. It begins with an overview that obesity is increasing globally and is associated with various health risks. It then discusses various factors that influence energy balance and can lead to obesity, including dietary intake, energy expenditure, physical activity, and psychosocial factors. Key hormones and brain regions such as the hypothalamus that regulate appetite and food intake are also examined. The document provides details on the causes and treatment of obesity.







![우리 뇌는 식욕을 어떻게 조절할까? [2016 대한청소년정신의학회 추계학술대회]](https://cdn.slidesharecdn.com/ss_thumbnails/20161013-170201014103-thumbnail.jpg?width=640&height=640&fit=bounds)








































































